Спинальная амиотрофия Верднига-Гоффмана представляет собой наследственное заболевание. На фоне патологии отмечается повреждение в двигательных нейронах. Спинальная амиотрофия Гофмана передается с неполовыми хромосомами. Далее подробнее рассмотрим заболевание, его клиническую картину и возможные терапевтические мероприятия для его устранения.
Терминология
Прежде чем говорить о том, как проявляет себя спинальная амиотрофия, ознакомимся с некоторыми понятиями. Разберем название патологии. Оно состоит из двух частей:
- Спинальная - слово указывает на локализацию нарушения. В данном случае речь идет об определенном элементе, находящемся в позвоночнике. Это одна из важнейших структур организма - спинной мозг.
- Амиотрофия - слово, включающее три части: "а" - нарушения, "мио" - мышца" и "трофия" - питание.
На основании этой информации можно понять значение названия патологии. Спинальная амиотрофия Верднига-Гофмана, таким образом, представляет собой нарушение питания в мускулатуре. Патология характеризуется наличием слабости и подергиванием волокон.
Наследование
Спинальная мышечная амиотрофия относится к аутосомно-рецессивным заболеваниям. Это определение указывает на тип наследования, при котором передача признака осуществляется посредством неполовых хромосом. При этом он проявляется только тогда, когда присутствует изначально у обоих родителей (сами они могут не болеть).

Развитие заболевания
Спинальная амиотрофия взрослых не встречается. Патология проявляет себя у детей. Недуг характеризуется злокачественным течением и быстрым прогрессированием. За координацию движений отвечают крупные клетки спинного мозга. Они также поддерживают тонус мускулатуры. При их повреждении развивается дисфункция мышц.
Врожденная форма
Спинальная амиотрофия имеет три формы. Они определяются в соответствии со временем проявления первых признаков и интенсивностью развития процесса. Врожденная форма может начаться еще во внутриутробном периоде. В данном случае отмечается ослабление шевеления плода на более поздних сроках беременности. При этом в начале дородового периода движения были в пределах нормы. Само разрешение беременности может оказаться патологическим. Зачастую уже в течение первых нескольких дней после рождения обнаруживаются выраженные парезы мускулатуры, сопровождающиеся снижением ее тонуса и ухудшением сухожильных рефлексов. Также могут отмечаться ретробульбарные (ранние) симптомы. Они проявляются слабым криком младенца и вялым сосанием. В ряде случаев наблюдается полная арефлексия. У ребенка могут быть выявлены фибрилляции в языке, гипомимия, понижение глотательного рефлекса. Спинальная амиотрофия сопровождается тахикардией. Зачастую патология сочетается с несколькими пороками развития, замедлением формирования психики. Спинальная амиотрофия отличается быстрым течением и заканчивается к 1-1,5 годам летальным исходом.

Ранняя форма
Она отличается более мягким течением, нежели врожденная. Ранняя детская форма считается классическим проявлением заболевания. Спинальная амиотрофия в этом случае проявляется в возрасте до полутора лет.
Практически во всех случаях признаки недуга обнаруживаются после пищевого отравления или какого-либо инфекционного поражения. Развивающийся нормально ребенок начинает достаточно быстро терять ранее приобретенные двигательные способности. Он перестает сидеть, стоять и ходить. Сначала отмечаются вялые парезы в нижних конечностях, постепенно переходящие на туловище и руки. Состояние ребенка ухудшается очень быстро. В мышцах шеи и бульбарной мускулатуре появляется слабость. В результате недостаточности дыхательной системы к 4-5 годам появляется пневмония, затем наступает смерть. Вялые парезы у детей осложняются сухожильными контрактурами. Зачастую спинальная амиотрофия Верднига-Гоффмана сопровождается общим гипергидрозом.
Позднее появление патологии
Третья форма заболевания начинается после 1,5-2 лет. По сравнению с предыдущими она протекает сравнительно легко. Способность к передвижению сохраняется у детей до 10 лет. После этого состояние, как правило, ухудшается.

Клиническая картина
Для патологии характерны парезы сначала проксимальных отделов нижних конечностей, а затем и верхних. При спинальной амиотрофии жировой подкожный слой хорошо выражен. Это, в свою очередь, затрудняет выявление дисфункции мускулатуры. Достаточно рано начинают угасать сухожильные рефлексы. Для патологии характерным является малый тремор пальцев при вытянутых руках. Типичными считаются деформации костей, в особенности нижних конечностей и грудины. Бульбарные симптомы проявляются в виде атрофии мускулатуры языка с подергиваниями фибриллярного типа, парезом в мягком небе и сниженным глоточным рефлексом.
Болезнь Фацио-Лонде
Это особый вариант проявления атрофии. Патология начинает развиваться, как правило, к трем годам жизни, и в некоторых случаях в подростковом возрасте. Для недуга характерна слабость мышц лица, в том числе жевательной мускулатуры. Отмечается затруднение глотания и изменения голоса. Патология сопровождается атрофией языка, в некоторых случаях может появиться офтальмоплегия. Прогрессирует болезнь очень быстро. Спустя 6-12 месяцев наступает смерть. К бульбарным нарушениям могут добавиться параличи и парезы в конечностях. В некоторых случаях эти симптомы не успевают даже развиться. Тем не менее, при вскрытии всегда выявляется поражение в клетках передних спинномозговых рогов на всем протяжении.

Диагностика
При проведении обследования патологию отделяют от миотонии Оппенгейма. Большинство специалистов считают, что эта патология не является самостоятельной нозологической единицей. Миотония Оппенгейма, по мнению исследователей, -синдром, для которого ведущим проявлением становится гипотония мышц выраженного типа. В этой связи в последнее время широко распространен термин "вялый ребенок".
Методы исследования: электромиография
Выявление спинальной амиотрофии базируется (кроме раннего проявления и типичной клинической картины) на результатах ряда дополнительных исследований. Из них стоит выделить электромиографию. Практически во всех случаях обнаруживается биоэлектрическая спонтанная активность в состоянии покоя при наличии потенциалов фасцикуляций. На фоне произвольных сокращений констатируется электрическая активность уреженного характера с ритмом "частокола". Это указывает на увеличение продолжительности потенциала и явления синхронизации.

Патоморфологическое исследование
Оно позволяет выявить уменьшение числа клеток передних спинномозговых рогов, а также изменения дегенеративного типа. Патологические нарушения резко выражены в районе шейного и поясничного утолщений, в двигательных ядрах черепных нервов. Обнаруживаются также изменения передних корешков, в интрамускулярных зонах нервных окончаний. Отмечается исчезновение и излишнее разветвление нормальных терминал.
Биохимический анализ
Это исследование позволяет выявить изменения углеводного обмена. Так, было обнаружено, что при спинальной амиотрофии гликолиз у пациентов приближен к эмбриональному типу. Достаточно часто выявляются существенные изменения креатин-креатининового обмена - повышение экскреции креатина, снижение выделения креатинина. Необходимо также отметить, что концентрация ферментов в сыворотке крови практически неизменна.

Спинальная амиотрофия: лечение
Терапия патологии сводится к назначению ЛФК и массажа. Эти процедуры должны выполняться регулярно. Радикальные методы лечения отсутствуют. В определенной степени облегчение может принести прием ряда медикаментов. В частности, специалисты рекомендуют такие средства, как "Сангвинарин", "Галантамин", "Оксазил", "Прозерин". Дополнительно назначаются витамины группы В. При выраженных проявлениях заболевания может быть рекомендовано повторное переливание крови в малых дозах.
Спинальная мышечная атрофия Верднига Гоффмана представляет собой редкое заболевание, которое возникает в нервно-мышечной системе человека. Данный недуг появляется вследствие различных мутаций. Медики считают, что основной причиной спинальной амиотрофии является плохая наследственность.
Признаки болезни будут кардинально различаться в зависимости от тяжести недуга. Медики условно разделяют спинально-мышечную атрофию на 4 степени тяжести. Отметим, что все 4 типа имеют общую особенность – у пациента происходят сбои в умственном развитии. на любых стадиях не может стать причиной дисфункции различных органов, расположенных в области таза.
Об атрофии мышц рук, ног и тела .
Главная причина возникновения болезни кроется в плохой генетике. Многочисленные научные исследования доказали, что атрофия спинного отдела появляется вследствие мутации в пятой хромосоме человека. Ген, который изменяется, ответственен за синтез специального белка, отвечающего за нормальное развитие двигательных нейронов. Вследствие того, что эти нейроны не могут в полном объеме выполнять все свои функции, у человека начинают атрофироваться мышцы тела.
Медики утверждают, что болезнь может появиться у ребенка только в том случае, если неправильный ген присутствует как у отца, так и у матери. Кроме того, врачи утверждают, что каждый второй человек на земле является носителем неправильного гена.
Симптомы при первой и второй форме болезни
Спинальная амиотрофия Верднига-Гофмана 1 типа чаще всего появляется в первые 6 месяцев жизни ребенка. В некоторых случаях заболевание можно обнаружить при диагностике во время родов. При 1 стадии у ребенка снижен тонус мышечных волокон, а рефлексы сухожилий могут вовсе отсутствовать. Иногда страдает функция глотания. Язык, как правило, тоже атрофируется. При 1 стадии у ребенка нарушается пищеварительная функция. Из-за этого еда может попадать в дыхательные пути. Дыхание тоже нарушено, из-за этого может появиться легочная недостаточность.Также у детей с спинальной амиотрофией 1 степени нарушается развитие моторики и наблюдается деформация грудной клетки. По статистике, если болезнь начинает прогрессировать сразу после рождения, ребенок погибает в первые 3-4 месяца жизни.
При спинальной атрофии 2 степени у ребенка появляются тревожные симптомы только спустя 6 месяцев после рождения. Изначально у пациента появляется атрофия мышц бедер. Сухожильные рефлексы постепенно притупляются. Мышцы лица, как правило, не подвержены атрофии. В некоторых случаях у больного возникают треморы в руках и нарушается дыхание. Мышцы шеи тоже со временем становятся слабыми. С течением времени у пациента появляется сколиоз или вывих тазобедренного сустава. Дети со спинальной атрофией 2 степени более подвержены воздействию патогенных инфекций из-за того, что дыхательная функция нарушена. При несвоевременном лечении пациент может погибнуть от удушья.
: причины и методы лечения недуга.
Что делать, если тянет поясницу? Подробнее о способах терапии .
Как правильно и эффективно делать ?
Симптоматика при третьем и четвертом типе амиотрофии
Спинальная амиотрофия Верднига-Гоффмана 3 стадии появляется, как правило, в юношеском возрасте. Характерный симптом при данном типе заболевания – неустойчивая ходьба. Объясняется это тем, что тонус мышц ног резко снижается. Это происходит из-за того, что мышечные волокна становятся тоньше. Пациент может часто спотыкаться или даже падать. С течением времени больной может и вовсе перестать ходить. Иногда атрофия появляется в мышцах рук. Мимика лица у пациента тоже нарушается. Скелет претерпевает изменения. У пациента деформируется грудная клетка и нарушается работа суставов. В редких случаях у пациента нарушается нормальное дыхание.
Спинальная амиотрофия Верднига-Гоффмана 4 стадии появляется, как правило, в зрелом возрасте. Характерный симптом у данной стадии недуга – возникновение слабости в ногах. Пациент может жаловаться на сильные боли в мышцах. С течением времени мышцы ног начинают атрофироваться, а рефлексы притупляться. Прогрессирование спинально-мышечной атрофии приводит к тому, что больной перестает ходить. При 4 степени дыхательная функция не изменяется. Мышцы рук работают полноценно. Медики считают, что 4 тип недуга является доброкачественным.
Диагностика и лечение патологии
Если у человека появились первые симптомы спинальной атрофии, ему назначают комплексную диагностику. Сначала пациент должен пройти электронейромиографию. С помощью этой процедуры медики могут обнаружить нарушения в работе нейронов. После этого больной должен пройти генетическое исследование. При проведении данной процедуры медики изучают данные, полученные из ДНК пациента. Генетическое исследование позволяет выявить мутации в 5 хромосоме.
Если у женщины есть близкие родственники, которые болели данным недугом, при беременности ей должны провести специальную пренатальную диагностику. Если у плода будет выявлена патология, может понадобиться принудительное прерывание беременности.
Спинальная атрофия не подвергается лечению. Однако можно облегчить состояние больного с помощью специальных препаратов, которые улучшают обменные процессы в нервных тканях. Терапию дополняют витаминами группы B, курсом анаболических стероидов, лечебной физкультурой и физиотерапией.
Дополнительные источники:
- 1.Su Y.N., Hung C.C., Lin S.Y., Chen F.Y., Chern J.P., Tsai C. screening for spinal muscular atrophy (SMA) // Public Library of Science
- 2. Sugarman E. A., Nagan N., Zhu H., Akmaev V.R., Zhou Z., Rohlfs E.M., Flynn K., Hendrickson B.C., Scholl T., Sirko-Osadsa D.A., Allitto B.A. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis European journal of human genetics.
- 3.Спинальная амиотрофия I, II, III, IV типа… // Центр молекулярной генетики Медико-генетического научного центра.


Из всех существующих и известных науке спинальных мышечных атрофий амиотрофия Верднига-Гоффмана является наиболее тяжелой разновидностью.
Распространенность этого заболевания находится сейчас на отметке 1 случай на 7-11 тыс. новорожденных малышей.
Ген, который вызывает это заболевание, имеется у каждого 50-го человека.
Однако, благодаря аутосомно-рецессивному типу наследования, проявляется нарушение у ребёнка только тогда, когда эта генетическая информация имеется у обоих родителей.
Поэтому вероятность того, что малыш родится с патологией, в данном случае составляет около 25%.
Можно ли справиться с таким заболеванием или хотя бы остановить прогрессию симптомов расскажем в этой статье.
Что такое амиотрофия Верднига-Гоффмана?
Спинальная амиотрофия 1-го типа или, по-другому, спинальная амиотрофия Верднига-Гоффмана - это особое заболевание нервной системы, передающееся по наследству (чаще всего от обоих родителей). Эта патология характеризуется наличием мышечной слабости практически во всей мышечной системы организма. Ребёнок, страдающий от такого заболевания, не может самостоятельно сидеть, передвигаться и обслуживать себя.
К большому сожалению в мире не существует лекарства от этого типа заболевания. Максимум, что могут предложить врачи в наше время - дородовая диагностика. Такое обследование помогает избежать рождения больного малыша в семье.
Свое название патология получила от двух учёных, впервые описавших её в конце 19 в. В настоящее время под понятием спинальной амиотрофии понимается несколько форм болезни, отличающихся клинически. Но все они при этом связаны одним и тем же генетическим дефектом, которым обладают родители ребёнка.
Клиническая картина заболевания
Спинальная амиотрофия имеет несколько форм и разновидностей , каждая из которых отличается возрастом появления характерных симптомов, тяжестью протекания заболевания и продолжительностью жизни пациентов.
Обычно эта патология приводит к инвалидности , поскольку нарушается двигательная система организма, и пациент не способен ни самостоятельно передвигаться, ни самостоятельно себя обслуживать. При тяжелых клинических ситуациях может понадобиться постоянный врачебный контроль в повседневной жизни.

Передвигаться такому больному помогают инвалидные кресла, ходунки, костыли, трости. К смертельному исходу такое заболевание может привести только в том случае, когда появляются осложнения со стороны дыхательной и сердечно-сосудистой системы (при пневмониях и сердечной недостаточностью).
Под воздействие патологии не попадают чувствительные нервные волокна , поэтому у ребёнка сохраняются все виды чувствительности. Не страдают также интеллект и ментальные функции, так при обучении ребёнок совершенно нормально воспринимает и усваивает информацию.
Классификация заболевания
В зависимости от возраста, при котором появились характерные симптому заболевания, амиотрофия Верднига-Гоффмана делится на несколько видов :
- Врожденная форма патологии . Примерный возраст появления изменений: от 0 до 6 месяцев. Обычно характеризуется слабым внутриутробным шевелением плода. При врождённой форме мышечная гипотония наблюдается с первых дней жизни малыша. В течение короткого времени происходит угасание глубоких рефлексов: ребёнок слабо кричит, плохо сосет молоко матери или соску, не может держать головку. Иногда случается так, что эти симптомы проявляются несколько позже, поэтому малыш может учиться держать головку и сидеть, но, поскольку имеется нарушение, эти навыки у него не разовьются. Также врожденная форма может сопровождаться бульбарными нарушениями, снижением глоточного рефлекса и фасцикулярными подергиваниями языка. Врожденная форма считается наиболее злокачественной и часто может сочетать в себе ещё и олигофрению, деформации грудной клетки, и 4 степени сколиоза . Быстрая обездвиженность и парез дыхательной системы приводит к дыхательной недостаточности и впоследствии к летальному исходу;
- Ранняя детская форма. При данной разновидности патологии первые симптомы могут проявиться после 6 месяцев. К этому моменту дети имеют нормальное физическое и психическое развитие. Они начинают потихоньку приобретать первые естественные навыки, вроде умения держать головку, стоять, садиться и переворачиваться. В большинстве случаев, при наличии данного типа заболевания, дети так и не научатся ходить. На начальной стадии возникают парезы в нижних конечностях, затем довольно быстро они развиваются в верхних конечностях и во всей мускулатуре. Наступает мышечная гипотония, угасают глубокие рефлексы, может проявиться тремор пальцев, непроизвольные мышечные сокращения. На более поздних этапах ко всем симптомам добавляются бульбарные нарушения, дыхательная недостаточность (прогрессирующая). Эта форма заболевание протекает медленнее, чем врожденный тип. Больные могут дожить вплоть до 15 лет;
- Амиотрофия Кугельберга-Веландера. Самая доброкачественная из всех форм спинальной амиотрофии. Симптомы проявляются после 2-х лет, иногда в период между 15-ю и 30-ю годами. При данной форме не встречается психической задержки развития, довольно длительное время пациенты способны двигаться самостоятельно. Многие доживают до глубокой старости на полном самообслуживании.
Факторы риска и причины заболевания
Поскольку спинальная амиотрофия Верднига-Гоффмана является наследственным заболеванием, то и причины её возникновения кроются в генетическом коде обоих родителей пациента . Проблема скрывается в пятой хромосоме, которая подвергается генетической мутации.
Мутирует ген, который отвечает за производство SMN белка . В здоровом организме синтез этого белка обеспечивает нормальное развитие двигательных нейронов. Если он подвергся мутации, то двигательные нейроны начинают разрушаться, что приводит к нарушению передачи импульса от нервного волокна к мышце. Как следствие - мышца не функционирует. Именно поэтому возникает двигательная атрофия и невозможность нормально передвигаться.

Ген с нарушениями имеет аутосомно-рецессивный тип наследования . Это значит, что для того, чтобы болезнь развилась, требуется совпадение двух мутировавших генов от обоих родителей. Т.е. по сути и отец, и мать должны быть носителями гена с патологией.
При этом они не больны, ведь у них имеется доминантный здоровый ген (это также обусловлено парностью генов). Если и у отца, и у матери малыша имеется ген с патологией, то риск того, что ребёнок родится с нарушениями составляет 25 %.
Видео: "Что такое спинальная мышечная атрофия?"
Методы диагностики заболевания
Когда речь заходит о диагностике такого рода заболевания, то нужно учесть, что для невролога, который будет проводить обследование, очень важен возраст появления первых симптомов у малыша.
Также важна динамика развития симптомов , данные неврологического статуса (т.е. наличие/отсутствие двигательных нарушений периферического типа на фоне нормальной чувствительности), наличие/отсутствие дополнительных врожденных аномалий и костных деформаций (склиоза , кифоза , лордоза , кривошеи).
Врожденный тип заболевания может быть выявлен неонатологом . Обследование проводится с миопатиями, мышечной дистрофией (прогрессирующей), боковым амиотрофическим склерозом, полиомиелитом, ДЦП и др. Если диагноз требует максимально точного подтверждения, то применяется также электронейромиография (исследования нервно-мышечного аппарата).
 Итоговый диагноз устанавливается только после получения данных о биологии мышц и исследования генетической ситуации
. Изучение ДНК-анализа позволяет генетикам выяснить гетерозиготное носительство генной аберрации (важно при планировании следующей беременности). Проводится также количественный анализ числа генов локуса СМА (позволяет высчитать наличие патологического гена у родителей.
Итоговый диагноз устанавливается только после получения данных о биологии мышц и исследования генетической ситуации
. Изучение ДНК-анализа позволяет генетикам выяснить гетерозиготное носительство генной аберрации (важно при планировании следующей беременности). Проводится также количественный анализ числа генов локуса СМА (позволяет высчитать наличие патологического гена у родителей.
Анализ ДНК, сделанный в дородовый период, может помочь снизить вероятность рождения ребёнка с патологией Вердинга-Гоффмана. Но сложность здесь заключается в том, что для получения ДНК-материала применяются инвазивные методы пренатальной диагностики (биопсия хориона, кордоцентез, амниоцентез).
Если внутриутробно болезнь подтвердится, то это выступит показанием к искусственному прерыванию текущей беременности.
Лечение заболевания
Современная медицина, к большому сожалению, ещё не разработала препараты, способные справиться с генетическими мутациями разного рода , поэтому лечебного курса, способного победить амиотрофию Верднига-Гоффмана, не существует. Есть ряд мероприятий, которые могут замедлить прогрессирующую атрофию мышц, но чудес ждать не приходится.
Медикаментозные препараты
Медикаменты способны только ослабить состояние больного , несколько поддержать и подпитать его нервную систему, но победить причину развития заболевания они не могу.
При спинальной амиотрофии применяют следующие препараты :
- Церебролизин, Цитофлавин, Глутаминовая кислота, АТФ, хлорид Карнитина, Метионин, Калия оротат и др. - эти препараты несколько улучшают метаболизм мышц и нервных тканей, поэтому их прописывают для периодического курсового приёма;
- Витамины В (Нейровитан, Мильгамма);
- Анаболические стероиды (Ретаболил, Неробол);
- Прозерин, Нейромидин, Дибазол - эти препараты способны несколько улучшить мышечную проводимость.

Упражнения, массаж, ЛФК
А знаете ли вы, что…
Следующий факт
Физические упражнения и массаж также применяются для облегчения состояния пациента . Однако, в случае с такой серьёзной патологией, ЛФК и массаж должны прописываться вашим лечащим врачом и вестись под строгим соблюдением специалиста (то же касается и физиотерапии). Никакие базовые комплексы ЛФК здесь применять нельзя, только индивидуально подобранные доктором в соответствии с уровнем развития патологии, характерными симптомами и стадией заболевания.
Видео: "Лечение спинальной мышечной атрофии с помощью генной терапии"
Лечение в домашних условиях
Самовольное лечение дома без консультации и наблюдения врача строго запрещается в данном случае . Профилактические меры дома могут быть применены только если на это дал разрешение доктор. Специалист может прописать вам какие-то несложные упражнения, которые вы сможете делать на дому. Если таких рекомендаций не было, то самостоятельно что-то делать строго запрещается.
Профилактика заболевания
Основываясь на том, что амиотрофия Вердинга-Гоффмана - это наследственное заболевание, то никаких профилактических мер для него не существует . К сожалению, если патология у малыша выявилась ещё в дородовый период, целесообразным будет прибегнуть к искусственному прерыванию неудачной беременности. Ни лечение, ни профилактика для такого рода заболеваний ещё не изобретены.
Прогноз заболевания
К несчастью, прогнозы для данной патологии крайне неблагоприятны . Шансов на выздоровление нет и не может быть. Если ребёнок родился с врожденной формой амиотрофии, то он погибнет в период между 6 месяцами и 2 годами жизни. Более поздние виды заболевания продлят больному жизнь, но вероятность летального исхода остается очень высокой.
Заключение
Современная медицина знает много заболеваний, с которыми не может справиться ни одна методика и ни один препарат. Спинальная амиотрофия Верднига-Хоффмана одна из них. Генетическое происхождение данной патологии сделало невозможным её лечение известными нам способами.
Также есть ещё несколько важных моментов, которые вам стоит учитывать, если у вас есть риск рождения малыша с данным заболеванием :
- Спинальная амиотрофия Верднига-Хоффмана имеет несколько разновидностей : врожденная, ранняя и амиотрофия Кугельберга-Веландера. Врожденная форма приводит к раннему летальному исходу (примерно в возрасте от 6 месяцев до 2-х лет), ранняя форма и поздняя могут продлить жизнь пациента, но шансов на выздоровление так или иначе нет;
- Риск развития патологии может появиться только при условии, что обоих родителей есть мутировавший ген . Только при условии, что и мать, и отец (которые при этом не выглядят больными, а являются лишь носителями гена) являются обладателями нарушений в генетической цепочке, может родиться ребёнок с заболеванием Верднига-Хоффмана (25% шанс);
- Вылечить генетическое заболевание невозможно, также как и невозможно провести профилактику . Медикаментозные средства прописываются только для облегчения симптомов пациента и для поддержания в организме тонуса. ЛФК и массаж прописываются только лечащим врачом, только им и никем другим, поскольку заболевание серьезное и требует строгого наблюдения. Если при дородовой диагностике уже был выявлен тот факт, что ребёнок родится с патологией, то врачи советуют прибегнуть к искусственному прерыванию беременности.
Пройдите тест!
Спинальная мышечная атрофия Верднига-Гоффмана – прогрессирующая нервно-мышечная патология, которая передается по наследству. Заболевание приводит к поражению проксимальной поперечнополосатой мускулатуры туловища, нижних конечностей и шеи.
Причины возникновения
Это наследственная патология, в основе которой лежит мутация в 5 человеческой хромосоме . Как результат нарушается синтез белка SMN, который требуется для нормального развития двигательных нейронов.
Болезнь приводит к разрушению или развитию неполноценных нервных клеток, не способных передать импульсы к мышечным волокнам. Поэтому иннервируемые мышцы перестают работать, развивается атрофия.
Согласно статистике, каждый второй человек является носителем патологического гена.
Для мутированного гена характерен аутосомно-рецессивный тип наследования – для развития заболевания потребуется совпадение двух неправильных хромосом от матери и отца.

Больной ребенок рождается только от родителей-носителей патологического гена. Однако мать и отец имеют здоровый доминантный ген, поэтому симптомы патологии у них не проявляются.
Клиническая картина заболевания
Симптоматика и тяжесть течения СМА определяется временем манифестации первых признаков заболевания. Поэтому медики выделяют 3 вида болезни:
- врожденную форму;
- раннюю детскую;
- позднюю форму.

Стоит детальнее рассмотреть каждую из форм.
Особенности врожденной формы
Характерно рождение детей с вялыми парезами. У новорожденных диагностируют генерализованную мышечную гипотонию, отсутствие глубоких рефлексов. Бульбарные расстройства приводят к тому, что малыш вяло сосет грудь, тихо кричит, у него снижен глоточный рефлекс. Со временем у ребенка диагностируют парез диафрагмальной мышцы.
Симптомы спинальной амиотрофии Верднига-Гоффмана проявляются у 1 ребенка из 10 тысяч новорожденных.
Заболевание нередко сочетается с деформациями костей и суставов (воронкообразной грудиной, сколиозом, контрактурой суставов). Характерно замедленное развитие статический и локомоторных функций, поэтому лишь ограниченное число малышей способны держать голову самостоятельно. Однако данный навык может быстро регрессировать.

Многие пациенты с врожденной формой спинальной миопатией имеют сниженный интеллект, пороки развития (косолапость, гидроцефалия, гемангиома, крипторхизм).
Болезнь развивается быстро, характеризуется злокачественным течением. Пациенты редко доживают до 9 лет. Причиной смерти становятся соматические патологии.
Более 50% детей, которые болеют врожденной формой амиотрофией Гоффмана-Верднига не доживают до 2 лет.
Симптомы ранней детской формы
Для синдрома характерно развитие первых признаков патологии у младенцев старше 6 месяцев. При этом у пациентов отмечается удовлетворительное моторное развитие: они способны держать головку, сидеть, иногда умеют стоять. Характерно подострое развитие болезни на фоне интоксикации или инфекционного заболевания.

Миопатия приводит к появлению вялых парезов ног, которые стремительно распространяются на мускулатуру туловища и рук. Это провоцирует снижение мышечного тонуса, глубоких рефлексов. На поздних стадиях у ребенка диагностируют генерализованную мышечную гипотонию, бульбарный паралич.
Прогноз у патологии неблагоприятный – болезнь имеет злокачественное течение, дети редко доживают до 15 лет.
Поздняя форма
Первые признаки патологии возникают после полного завершения формирования локомоторных и статических функций. Поэтому многие дети умеют самостоятельно бегать и ходить. Характерно постепенно развитие миопатии, которая проявляется в неловких и неуверенных движениях ребенка. Постепенно начинает меняться походка – дети ходят, как заводные куклы, постоянно сгибая ноги в коленях.

Вялый парез первоначально локализован в нижних конечностях, но постепенно распространяется на мускулатуру туловища и рук. Ребенок имеет хорошо развитую подкожно-жировую клетчатку, поэтому атрофия мышечных волокон малозаметна. Постепенно у пациента появляется бульбарная симптоматика, возникает тремор пальцев.
На ранних стадиях СМА отмечается угасание глубоких рефлексов, начинает деформироваться грудная клетка.
Болезнь имеет злокачественное течение, однако симптоматика развивается медленнее, чем у предыдущих форм. Дети теряют способность к самостоятельному передвижению лишь к 10 годам. Смерть наступает обычно до 30 лет.
| Тип СМА | Манифест заболевания | Максимальная функция | Возраст наступления смерти |
| Врожденная форма | Первые симптомы развиваются у детей до 6 месяцев | Ребенок не способен двигаться, держать голову, сидеть | Многие пациенты умирают раньше 2 лет, но могут дожить до 9 лет |
| Ранняя детская форма | Симптоматика возникает с 7 до 12 месяцев | Пациент может сидеть, стоять, однако постепенно функции регрессируют | 14-15 лет |
| Поздняя форма | Признаки заболевания развиваются у детей старше 1 года | Ребенок стоит и ходит | Возраст от 20 до 30 лет |
Диагностические мероприятия
Для постановки диагноза большое значение имеет возраст появления первых симптомов патологии, динамика развития и неврологический статус пациента (нарушения двигательных функций на фоне сохранения чувствительности), наличие костной деформации и врожденных аномалий развития. Врожденную форму СМА обычно диагностируют неонатологи.
Комплексная диагностика предполагает проведение следующих мероприятий:

Чтобы снизить риск рождения ребенка с СМА, рекомендуют проводить дородовое исследование ДНК. Однако получить материал для диагностики можно только посредством инвазивных методик (амниоцентеза, биопсии хориона, кордоцентеза). Если амиотрофия была диагностирована внутриутробно, то показано прерывание беременности.

Особенности терапии заболевания
Спинальная миопатия является неизлечимой патологией, поэтому лечение позволяет лишь облегчить состояние пациента. С этой целью применяют такие препараты:
- анаболические стероиды;
- витамины группы В;
- средства, улучшающие метаболизм мускулатуры и нейронов;
- препараты, улучшающие нервно-мышечную проводимость.
Дополнительно назначают курс массажа и ЛФК , физиотерапию (электростимуляцию мускулатуры, оксигенотерапию), ортопедическую коррекцию. Пациент должен придерживаться диетического питания.

Если развивается дыхательная недостаточность, то больного ребенка подключают к аппарату искусственной вентиляции легких, чтобы восстановить дыхание. При выраженном нарушении глотательного рефлекса показано использование гастростомической питательной трубки. Большинству пациентов с СМА приходится передвигаться на инвалидной коляске.
Ученые из различных стран мира проводят научные исследования, чтобы создать препарат, способный повышать продукцию белка SMN. Однако на данный момент научная работа не принесла желаемого результата.
Болезнь не имеет специфических методов профилактики. Снизить риск развития патологий у будущего ребенка позволит лишь консультация с генетиком на стадии планирования беременности. При необходимости проводится генетическое исследование, чтобы определить наличие патологического гена у родителей.
0Частота заболевания
СМА – одно из самых часто встречеющихся заболеваний из орфанных (редких), болеет один новорожденный на 6000-10000 .
Причина СМА
СМА — наследственное заболевание, оно связано с мутациями в гене SMN1.
Чтобы болезнь проявилась, носителями мутации в этом гене должны быть оба родителя. Рецессивный ген СМА имеет примерно каждый 40-й. Вероятность рождения больного ребенка от двух носителей – 25%, с такой же вероятностью ребенок двух носителей не будет иметь генной поломки. Ещё в 50% случаев он будет носителем СМА, но сам не заболеет.
В редких случаях (менее 2%) больные дети рождаются в семьях, где носителем является только один родитель. У второго родителя мутация гена происходит при закладке яйцеклетки или сперматозоида.
Что повреждается в результате мутации
Из-за дефектного гена в организме нарушается выработка белка SMN — протеина выживаемости мотонейронов. Без этого белка мотонейроны – нервные клетки спинного мозга, отвечающие за координацию движений и мышечный тонус – отмирают, сигнал в мышцы ног, спины и отчасти рук не идёт.
Без необходимого тонуса мышцы постепенно атрофируются. Отсутствие мышц пресса и спины приводит, кроме прочего, к обширным искривлениям позвоночника, а они – к проблемам с дыханием, которые из-за слабых мышц и так есть.
Болезнь может проявляться с первых месяцев жизни или в более позднем возрасте.

От чего зависит степень тяжести болезни
За выработку белка SMN отвечают два гена — SMN1 и SMN2.
При этом SMN1 – основной «заказчик» данного белка, а SMN2 – дополнительный, он вырабатывает белок в количестве, недостаточном для нормальной работы организма. В случаях, когда в геноме человека SNM1 отсутствует, SNM2 начинает выполнять замещающие функции, но никогда не может полностью восполнить недостачу.
Копий SMN2 в геноме бывает до восьми. От имеющегося у человека числа копий SMN2 и зависит тяжесть состояния больного. Такой сложный механизм болезни приводит к тому, что СМА имеет несколько форм, и состояние больных – очень разное.
Какие формы СМА существуют?
Существует 4 типа СМА, различающиеся степенью тяжести и возрастом, в котором впервые проявляется заболевание.
СМА I, болезнь Верднига-Гоффмана. Самая тяжёлая форма болезни, проявляется у младенцев от 0 до 6 месяцев. Дети с этой формой с рождения имеют трудности с дыханием, сосанием и глотанием, а также не осваивают самые простые контролируемые движения — не держат голову, не сидят самостоятельно. Ранее считалось, что большинство (80%) не доживают до двух лет. Сейчас благодаря новым стратегиям ИВЛ и зондовому кормлению срок жизни можно продлить ещё на несколько месяцев.
СМА II, болезнь Дубовица. Первые проявления болезни в 7-18 месяцев. Человек с таким типом СМА может есть и сидеть, но не ходит самостоятельно. Продолжительность жизни зависит от степени поражения мышц, обеспечивающих дыхание.
СМА III , болезнь Кюгельберга-Веландер. Болезнь впервые проявляется после полутора лет. Такие больные могут стоять (испытывая боль), но не ходят. На продолжительность жизни СМА III типа, как правило, не влияет, но сильно ухудшает её качество.
СМА
IV
, этот тип называется ещё «взрослой СМА»,
поскольку болезнь проявляется обычно в возрасте после 35 лет.
Симптомы – мышечная слабость, сколиоз и тремор. Кроме того, развиваются контрактуры суставов (ограничения подвижности в суставах) и нарушения метаболизма.
Прогрессирование заболевания не очень быстрое, сначала мышечная слабость затрагивает мышцы ног, затем – рук. Обычно проблем с глотательной и дыхательной функцией у больных нет.
Большинство из больных IV типом СМА могут ходить, и лишь некоторым приходится прибегать к инвалидным коляскам.
СМА, связанные с нарушением гена SMN, в медицинской литературе называют проксимальными — они составляют 95% от всех спинальных амиотрофий. СМА, не связанных с геном SMN, довольно много, но встречаются они редко. К ним относится, например, болезнь Кеннеди. Исследования 1990-х годов показали, что болезнь Кеннеди не связана не с поломкой гена SMN1, но с другими генетическими мутациями, приводящими к нарушению усвоения белка SMN. Болезнь проявляется у людей старше 35 лет. Для СМБА характерна, в основном, слабость конечностей.
Один из видов СМА, не связанный с геном SMN, называется болезнь Кеннеди. То, что эту болезнь до сих пор иногда относятся к СМА – анахронизм. В конце 1960-х, когда было выполнено подробное описание этой атрофии, её посчитали разновидностью СМА, так как при ней поражаются те же нервы и мышцы, что и при трёх типах СМА (но в гораздо меньшей степени).
Как это лечат?
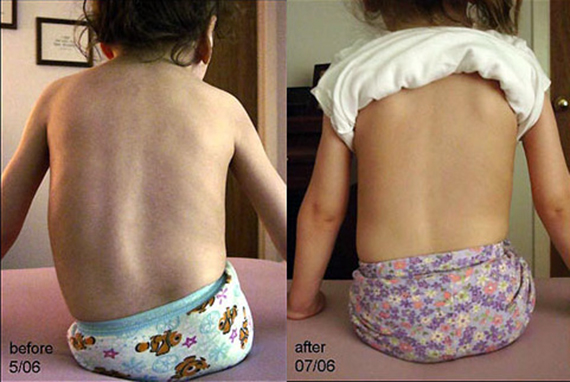
На сегодняшний момент радикального лечения от СМА не существует.
Международной корпорацией «Биоген» был разработан препарат «Спинраза», который значительно улучшил состояние больных, к которым применялся во время тестирования. В настоящее время препарат одобрен к применению в США, в Европе ориентировочная стоимость годового курса, по подсчётам компании, будет составлять около 270 тысяч евро, в России препарат не сертифицирован. Лечение пожизненное.
Можно ли помочь больным СМА и как именно?
Вылечить болезнь пока нельзя, но можно облегчать состояние больных СМА, то есть различными способами компенсировать проявления болезни.
При тяжёлых типах СМА больным приходится помогать дышать и глотать. Поэтому им жизненно необходимы мобильные аппараты ИВЛ, аспираторы-откашливатели, мешки Амбу.
Ещё детям со СМА очень нужна помощь волонтёров, способных хоть на короткое время подменить родителей.
Детям, больным СМА, помощь может понадобиться в любой момент, поэтому мамы и папы всегда начеку и сами осваивают навыки реанимации, необходимые на случай, если ребенок внезапно перестал дышать.
Менее тяжёлым больным нужны лекарства, облегчающие дыхание, корсеты, коляски и другие приспособления, облегчающие перемещение и жизнь людей со слабыми мышцами.
Болезнь, продолжающаяся много лет, выматывает, поэтому пациентам, особенно взрослым, часто необходима помощь психолога.

Фонд работает по всей территории России. Работа фонда имеет два основных направления — оказание помощи самим больным СМА и их близким и работа на системные изменения ситуации со СМА в России.
Вы можете поддержать деятельность фонда, сделав пожертвование любым удобным для вас способом. Помочь можно, оформив разовое или регулярное пожертвование на специальной странице фонда или отправив на короткий номер 3443 смс со словом СМА и, через пробел, суммой пожертвования — например, СМА 300 .
Можно ли заболеть СМА из-за прививок?
В Европе и США связь между прививками и проявлением болезни не прослежена.
Понять, есть ли связь между СМА и прививками, может объяснение разницы между СМА и полиомиелитом. Полиомиелит – инфекционное заболевание, когда от инфекции повреждается организм изначально здорового ребёнка. Ребёнок со СМА, родившийся с повреждённым геномом, внешне может выглядеть здоровым, но на самом деле он уже болен, просто симптомы его болезни проявляются постепенно. В этом отношении СМА – такая же «отложенная» болезнь как, например, миодистрофия Дюшенна или синдром Ретта, когда ребёнок, некоторое время развивавшийся в соответствии с нормой, теряет приобретённые ранее навыки и становится инвалидом.
Большинство проявлений СМА связаны с освоением первых двигательных навыков. Первые проявления болезни совпадают по времени с несколькими возрастными прививками. В итоге человек и его родные могут утверждать, что он «заболел от прививки», но на самом деле у него просто проявились признаки болезни, которая уже была.
Как определяют, что у ребенка именно СМА, а не какая-то другая болезнь?
Несмотря на то, что впервые СМА была описана австрийским неврологом Гвидо Верднигом и немецким неврологом Джоханном Хоффманном ещё в начале 1890-х годов, полностью понять природу заболевания удалось только в конце XX века. Ген SMN1 был открыт в 1995 году. Чтоб подтвердить диагноз СМА, нужен генетический тест.
В России соответствующие генетические тесты стали доступны в начале 2000-х годов. Генетический тест на СМА возможно сделать по ОМС, однако на практике не слишком много врачей знают этот редкий диагноз и направляют больных на соответствующее исследование. Стоимость такого тестирования в коммерческих лабораториях Москвы – порядка 6 тысяч рублей.
Отсутствие специальной диагностики привело также к путанице в диагнозах. Большинство больных СМА в России не выявлены, у многих выявленных в качестве диагноза записана «болезнь Верднига-Гоффмана», хотя не у всех из них (особенно взрослых) в действительности именно этот тип болезни.
Сколько больных СМА в России?

Препарат «Спинраза», значительно улучшающий состояние больных. Фото с сайта healthbeat.spectrumhealth.org
С учётом частоты заболевания, количество больных СМА в России должно составлять от семи до двадцати четырёх тысяч человек. На сегодняшний день в реестре пациентов фонда «Семьи СМА» находится около 400 человек.
Кто в России помогает людям со СМА и их семьям
Благотворительный фонд «Вера», детский хоспис «Дом с маяком», благотворительный фонд «Детский паллиатив», благотворительный фонд «Семьи СМА», детская паллиативная служба «Милосердие».
С 2014 года в Москве развивается совместный проект службы «Милосердие» и фонда «Семьи СМА» «Клиники СМА» На встречах, которые проходят раз в месяц, больные могут получить консультации пульмонолога, ортопеда, физиотерапевта и психолога. В последнее время часть встреч ориентированы и на нужды взрослых пациентов.
Известные люди со СМА

Итальянка Симона Спиноглио
родилась с наследственным заболеванием – спинальной мышечной атрофией 2 типа. Она с самого рождения не может ходить и передвигается только с помощью электрической коляски. Но ее жизнь полна и насыщенна; ничто не может помешать ее стремлению жить.
Симона работает на «горячей линии» итальянской Ассоциации «Семьи СМА» (Famiglies of SMA) и помогает детям и взрослым со СМА и другими нервно-мышечными заболеваниями.
Также Симона записала несколько популярных в итальянском сообществе СМА песен — о свободе делать то, что ты хочешь, несмотря на болезнь.
Российская певица Юлия Самойлова родилась в городе Ухта (Республика Коми) В возрасте десять лет выступила на благотворительном концерте, после чего была приглашена заниматься пением в местный Дворец пионеров. В пятнадцати лет начала заниматься в городском Доме культуры.
В 2008 году собрала собственную музыкальную группу (распалась в 2010). В 2013 году приняла участие в конкурсе «Фактор А» на телеканале «Россия». Заняла второе место и получила персональную премию Аллы Пугачёвой «Золотая звезда Аллы». В 2017 году из-за недопуска России в конкурсную программу не смогла принять участие в конкурсе «Евровидение». Передвигается на коляске.
Программист из Владимира Валерий Спиридонов . Окончил школу с золотой медалью, затем защитил диплом инженера. В 2015 году Валерий планировал стать участником эксперимента итальянского хирурга Серджио Канаверо по пересадке головы человека (эксперимент был отменен).
Сегодня Валерий — член городской общественной палаты Владимира, эксперт по вопросам доступной среды, а также создатель собственного сообщества «Desire for life», рассказывающего о создании доступной среды и перспективных медицинских проектах. Валерий – участник многих телепрограмм на российском и зарубежном ТВ.