Do cytowania: Leonova M.V. Nowe i obiecujące leki blokujące układ renina-angiotensyna-aldosteron // RMJ. Opinia medyczna. 2013. nr 17. 886
Obecnie uważa się, że rola układu renina-angiotensyna-aldosteron (RAAS) w rozwoju nadciśnienia tętniczego (NT) i innych chorób układu krążenia jest dominująca. W kontinuum sercowo-naczyniowym nadciśnienie jest jednym z czynników ryzyka, a głównym patofizjologicznym mechanizmem uszkodzenia układu sercowo-naczyniowego jest angiotensyna II (ATII). ATII jest kluczowym składnikiem RAAS - efektorem realizującym skurcz naczyń, retencję sodu, aktywację współczulnego układu nerwowego, proliferację i hipertrofię komórek, rozwój stresu oksydacyjnego i stanu zapalnego ściany naczyń.
Obecnie opracowano i szeroko stosuje się klinicznie dwie klasy leków blokujących RAAS – inhibitory ACE i blokery receptora ATII. Efekty farmakologiczne i kliniczne tych klas są różne. ACE jest peptydazą metaloproteinazy cynkowej, która metabolizuje ATI, AT1-7, bradykininę, substancję P i wiele innych peptydów. Mechanizm działania inhibitorów ACE związany jest głównie z zapobieganiem powstawaniu ATII, co sprzyja wazodylatacji, natriurezie i niweluje prozapalne, proliferacyjne i inne efekty ATII. Ponadto inhibitory ACE zapobiegają degradacji bradykininy i podwyższają jej poziom. Bradykinina jest silnym środkiem rozszerzającym naczynia krwionośne, nasila natriurezę, a co najważniejsze ma działanie kardioprotekcyjne (zapobiega przerostowi, zmniejsza uszkodzenia niedokrwienne mięśnia sercowego, poprawia ukrwienie wieńcowe) oraz wazoprotekcyjne, poprawiając funkcję śródbłonka. Jednocześnie wysoki poziom bradykininy jest przyczyną rozwoju obrzęku naczynioruchowego, co jest jedną z poważnych wad inhibitorów ACE, znacznie podwyższających poziom kinin.
Inhibitory ACE nie zawsze są w stanie całkowicie zablokować tworzenie się ATII w tkankach. Obecnie ustalono, że inne enzymy, które nie są związane z ACE, przede wszystkim endopeptydazy, na które inhibitory ACE nie mają wpływu, mogą również uczestniczyć w jego przemianie w tkankach. W rezultacie inhibitory ACE nie mogą całkowicie wyeliminować działania ATII, co może być przyczyną ich braku skuteczności.
Rozwiązanie tego problemu ułatwiło odkrycie receptorów ATII i pierwszej klasy leków, które selektywnie blokują receptory AT1. Poprzez receptory AT1 realizowane są niekorzystne efekty ATII: zwężanie naczyń, wydzielanie aldosteronu, wazopresyny, norepinefryny, zatrzymywanie płynów, proliferacja komórek mięśni gładkich i kardiomiocytów, aktywacja SAS, a także mechanizm ujemnego sprzężenia zwrotnego – tworzenie reniny . Receptory AT2 pełnią „korzystne” funkcje, takie jak rozszerzanie naczyń, procesy naprawcze i regeneracyjne, działanie antyproliferacyjne, różnicowanie i rozwój tkanek embrionalnych. W działaniu klinicznym blokerów receptora ATII pośredniczy eliminacja „szkodliwego” działania ATII na poziomie receptorów AT1, co zapewnia pełniejsze blokowanie działań niepożądanych ATII i nasilenie działania ATII na receptory AT2 , co uzupełnia działanie wazodylatacyjne i antyproliferacyjne. Blokery receptora ATII mają specyficzny wpływ na RAAS bez ingerencji w układ kininowy. Brak wpływu na czynność układu kininowego z jednej strony zmniejsza nasilenie działań niepożądanych (kaszel, obrzęk naczynioruchowy), z drugiej pozbawia blokery receptora ATII ważnego działania przeciwniedokrwiennego i wazoprotekcyjnego, co odróżnia je od inhibitorów ACE. Z tego powodu wskazania do stosowania blokerów receptora ATII w większości powtarzają wskazania do wyznaczania inhibitorów ACE, czyniąc je lekami alternatywnymi.
Pomimo wprowadzenia blokerów RAAS do powszechnej praktyki leczenia nadciśnienia tętniczego, problemy z poprawą wyników leczenia i rokowania nadal pozostają. Należą do nich: możliwość poprawy kontroli ciśnienia tętniczego w populacji, skuteczność leczenia nadciśnienia tętniczego opornego, możliwość dalszego zmniejszenia ryzyka chorób układu krążenia.
Aktywnie trwają poszukiwania nowych sposobów wpływania na RAAS; badane są inne ściśle oddziałujące układy i opracowywane są leki o wielu mechanizmach działania, takie jak inhibitory ACE i neutralnej endopeptydazy (NEP), enzymu konwertującego endotelinę (EPF) i inhibitory NEP, inhibitory ACE/NEP/EPF.
Inhibitory wazopeptydazy
Oprócz dobrze znanego ACE, wazopeptydazy obejmują 2 inne metaloproteinazy cynku – neprylizynę (neutralną endopeptydazę, NEP) oraz enzym konwertujący endotelinę, które również mogą być celem efektów farmakologicznych.
Neprylizyna jest enzymem wytwarzanym przez śródbłonek naczyń i bierze udział w degradacji peptydu natriuretycznego, a także bradykininy.
Układ peptydów natriuretycznych jest reprezentowany przez trzy różne izoformy: przedsionkowy peptyd natriuretyczny (typ A), mózgowy peptyd natriuretyczny (typ B), które są syntetyzowane w przedsionku i mięśniu sercowym oraz śródbłonkowy peptyd C, które są endogennymi inhibitorami RAAS w ich funkcje biologiczne oraz endotelina-1 (Tabela 1). Sercowo-nerkowe działanie peptydu natriuretycznego polega na obniżeniu ciśnienia krwi poprzez wpływ na napięcie naczyń i gospodarkę wodno-elektrolitową, a także działanie antyproliferacyjne i przeciwzwłóknieniowe na narządy docelowe. Ostatnio układ peptydów natriuretycznych jest zaangażowany w metaboliczną regulację utleniania lipidów, tworzenie i różnicowanie adipocytów, aktywację adiponektyny, wydzielanie insuliny i tolerancję węglowodanów, co może zapewniać ochronę przed rozwojem zespołu metabolicznego.
Do chwili obecnej wiadomo, że rozwój chorób sercowo-naczyniowych związany jest z dysregulacją układu peptydów natriuretycznych. Tak więc w nadciśnieniu występuje niedobór peptydu natriuretycznego, co prowadzi do wrażliwości na sól i upośledzonej natriurezy; w przewlekłej niewydolności serca (CHF) na tle niedoboru obserwuje się nieprawidłowe funkcjonowanie hormonów układu peptydów natriuretycznych.
Dlatego też inhibitory NEP mogą być stosowane do wzmocnienia układu peptydów natriuretycznych w celu uzyskania dodatkowego działania hipotensyjnego i ochronnego na układ sercowo-nerkowy. Hamowanie neprylizyny prowadzi do nasilenia działania natriuretycznego, moczopędnego i wazodylatacyjnego endogennego peptydu natriuretycznego, aw rezultacie do obniżenia ciśnienia krwi. Jednak NEP bierze również udział w degradacji innych peptydów wazoaktywnych, w szczególności ATI, ATII i endoteliny-1. Dlatego bilans wpływu na napięcie naczyniowe inhibitorów NEP jest zmienny i zależy od przewagi działania zwężającego i rozszerzającego. Przy długotrwałym stosowaniu przeciwnadciśnieniowe działanie inhibitorów neprylizyny jest słabo wyrażone z powodu kompensacyjnej aktywacji tworzenia ATII i endoteliny-1.
Pod tym względem połączenie działania inhibitorów ACE i NEP może znacząco nasilać efekty hemodynamiczne i antyproliferacyjne w wyniku komplementarnego mechanizmu działania, co doprowadziło do powstania leków o podwójnym mechanizmie działania, zjednoczonych pod nazwą - inhibitory wazopeptydazy (tabela 2, ryc. 1).
Znane inhibitory wazopeptydaz charakteryzują się różnym stopniem selektywności względem NEP/ACE: omapatrilat - 8,9:0,5; fazidoprylat - 5,1:9,8; sampatrilat - 8,0:1,2. Dzięki temu inhibitory wazopeptydazy uzyskały znacznie większe możliwości uzyskania efektu hipotensyjnego, niezależnie od aktywności RAAS i poziomu retencji sodu oraz w ochronie narządów (regresja przerostu, albuminurii, sztywności naczyń). Najczęściej badanym w badaniach klinicznych był omapatrilat, który wykazywał wyższą skuteczność hipotensyjną w porównaniu z inhibitorami ACE, a u pacjentów z CHF prowadził do zwiększenia frakcji wyrzutowej i poprawy wyników klinicznych (badania IMPRESS, OVERTURE), ale bez przewagi nad inhibitorami ACE.
Jednak w dużych badaniach klinicznych z zastosowaniem omapatrilatu stwierdzono większą częstość występowania obrzęku naczynioruchowego w porównaniu z inhibitorami ACE. Wiadomo, że częstość występowania obrzęku naczynioruchowego podczas stosowania inhibitorów ACE wynosi od 0,1 do 0,5% w populacji, z czego 20% przypadków zagraża życiu, co wiąże się z wielokrotnym wzrostem stężenia bradykininy i jej metabolitów. Wyniki dużego wieloośrodkowego badania OCTAVE (n=25 302), które zostało specjalnie zaprojektowane do badania częstości występowania obrzęku naczynioruchowego, wykazały, że częstość występowania tego działania niepożądanego podczas leczenia omapatrilatem jest większa niż w grupie enalaprylu – 2,17% vs. 0,68% (ryzyko względne 3,4). Tłumaczono to zwiększonym wpływem na poziom kinin podczas synergistycznego hamowania ACE i NEP, związanego z hamowaniem aminopeptydazy P, która bierze udział w degradacji bradykininy.
Nowy podwójny inhibitor wazopeptydazy blokujący ACE/NEP, ilepatril, ma większe powinowactwo do ACE niż do NEP. Badając farmakodynamiczny wpływ ilepatrilu na aktywność RAAS i peptydu natriuretycznego u zdrowych ochotników stwierdzono, że lek w sposób zależny od dawki (w dawkach 5 i 25 mg) i znacząco (ponad 88%) hamuje ACE w osoczu przez ponad 48 godzin, niezależnie od wrażliwości na sól. Jednocześnie lek istotnie zwiększał aktywność reninową osocza przez 48 godzin i obniżał poziom aldosteronu. Wyniki te wykazały wyraźną i dłuższą supresję RAAS, w przeciwieństwie do inhibitora ACE ramiprylu w dawce 10 mg, co tłumaczono bardziej znaczącym tkankowym działaniem ilepatrylu na ACE i większym powinowactwem do ACE oraz porównywalnym stopniem blokady RAAS w porównaniu z połączeniem irbesartanu 150 mg + 10 mg ramiprylu. W przeciwieństwie do wpływu na RAAS, wpływ ilepatrilu na peptyd natriuretyczny objawiał się przejściowym zwiększeniem poziomu jego wydalania w okresie 4-8 godzin po podaniu dawki 25 mg, co wskazuje na mniejsze i słabsze powinowactwo dla NEP i odróżnia go od omapatrilat. Ponadto w zakresie wydalania elektrolitów lek nie wykazuje dodatkowego działania natriuretycznego w porównaniu z ramiprylem czy irbesartanem, a także innymi inhibitorami wazopeptydazy. Maksymalne działanie hipotensyjne występuje po 6-12 godzinach od przyjęcia leku, a obniżenie średniego ciśnienia krwi wynosi 5±5 i 10±4 mm Hg. odpowiednio przy niskiej i wysokiej wrażliwości na sól. Zgodnie z właściwościami farmakokinetycznymi, ilepatril jest prolekiem z aktywnym metabolitem, który szybko powstaje z maksymalnym stężeniem w ciągu 1-1,5 godziny i jest powoli eliminowany. Obecnie trwają badania kliniczne III fazy.
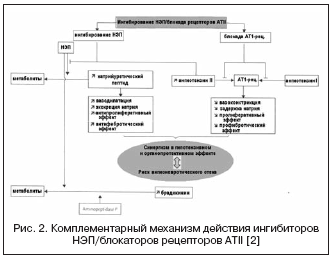
Alternatywną drogą podwójnej supresji RAAS i NEP jest połączenie blokady receptorów ATII i NEP (ryc. 2). Blokery receptora ATII nie wpływają na metabolizm kinin, w przeciwieństwie do inhibitorów ACE, dlatego potencjalnie mają mniejsze ryzyko rozwoju obrzęku naczynioruchowego. Obecnie pierwszy lek, bloker receptora ATII o działaniu hamującym NEP w stosunku 1:1, LCZ696, przechodzi III fazę badań klinicznych. Złożona cząsteczka leku zawiera walsartan i inhibitor NEP (AHU377) w postaci proleku. W dużym badaniu u pacjentów z nadciśnieniem tętniczym (n=1328) LCZ696 w dawkach 200-400 mg wykazał przewagę działania hipotensyjnego nad walsartanem w dawkach 160-320 mg w postaci dodatkowego obniżenia ciśnienia krwi o 5 /3 i 6/3 mmHg. . Hipotensyjnemu działaniu LCZ696 towarzyszyło wyraźniejsze obniżenie ciśnienia tętna: o 2,25 i 3,32 mm Hg. w dawkach odpowiednio 200 i 400 mg, co obecnie uważa się za pozytywny czynnik prognostyczny wpływu na sztywność ścian naczyń i wyniki sercowo-naczyniowe. Jednocześnie badanie biomarkerów neurohumoralnych podczas leczenia LCZ696 wykazało wzrost poziomu peptydu natriuretycznego przy porównywalnym stopniu wzrostu poziomu reniny i aldosteronu w porównaniu z walsartanem. Tolerancja u pacjentów z nadciśnieniem tętniczym była dobra, nie odnotowano przypadków obrzęku naczynioruchowego. Badanie PARAMOUMT zostało zakończone z udziałem 685 pacjentów z CHF i niezaburzoną EF. Wyniki badania wykazały, że LCZ696 szybciej i wyraźniej obniża poziom NT-proBNP (pierwszorzędowy punkt końcowy jest markerem zwiększonej aktywności peptydu natriuretycznego i złego rokowania w CHF) w porównaniu z walsartanem, a także zmniejsza wielkość lewego przedsionka , co wskazuje na regres jej przebudowy . Trwa badanie z udziałem pacjentów z CHF i obniżoną EF (badanie PARADIGM-HF).
Inhibitory układu endoteliny
Układ endoteliny odgrywa ważną rolę w regulacji napięcia naczyniowego i regionalnego przepływu krwi. Spośród trzech znanych izoform endotelina-1 jest najbardziej aktywna. Oprócz znanego działania wazokonstrykcyjnego, endotelina stymuluje proliferację i syntezę macierzy międzykomórkowej, a także poprzez bezpośredni wpływ na napięcie naczyń nerkowych bierze udział w regulacji homeostazy wodno-elektrolitowej. Działanie endoteliny jest realizowane poprzez interakcję z określonymi receptorami typu A i typu B, których funkcje są wzajemnie przeciwne: zwężenie naczyń następuje przez receptory typu A, a rozszerzenie naczyń przez receptory typu B. W ostatnich latach ustalono, że receptory typu B odgrywają ważną rolę w klirensie endoteliny-1, tj. blokada tych receptorów zaburza zależny od receptora klirens endoteliny-1 i zwiększa jej stężenie. Ponadto receptory typu B biorą udział w regulacji nerkowego działania endoteliny-1 oraz utrzymaniu ważnej homeostazy wodno-elektrolitowej.
Obecnie udowodniono rolę endoteliny w rozwoju wielu chorób, m.in. AH, CHF, nadciśnienie płucne, przewlekła choroba nerek; wskazuje na ścisły związek między poziomem endoteliny a zespołem metabolicznym, dysfunkcją śródbłonka i aterogenezą. Od lat 90 trwają poszukiwania antagonistów receptora endoteliny nadających się do użytku klinicznego; Znanych jest już 10 leków („sentanów”) o różnym stopniu selektywności względem receptorów typu A/B. Pierwszy nieselektywny antagonista receptora endoteliny – bozentan – w badaniu klinicznym u pacjentów z nadciśnieniem wykazał skuteczność hipotensyjną porównywalną do inhibitora ACE, enalaprylu. Dalsze badania nad skutecznością antagonistów endoteliny w nadciśnieniu wykazały ich znaczenie kliniczne w leczeniu nadciśnienia tętniczego opornego i obciążonego dużym ryzykiem sercowo-naczyniowym. Dane te uzyskano w dwóch dużych badaniach klinicznych DORADO (n=379) i DORADO-AC (n=849), w których darusentan dodano do trójlekowej terapii skojarzonej u pacjentów z opornym nadciśnieniem tętniczym. W badaniu DORADO pacjenci z nadciśnieniem opornym byli związani z przewlekłą chorobą nerek i białkomoczem, a w wyniku dodania darusentanu zaobserwowano nie tylko znaczny spadek ciśnienia krwi, ale także zmniejszenie wydalania białka. Przeciwbiałkomoczowe działanie antagonistów receptora endoteliny zostało następnie potwierdzone w badaniu z udziałem pacjentów z nefropatią cukrzycową z zastosowaniem awocentanu. Jednak w badaniu DORADO-AC nie stwierdzono przewagi dodatkowej redukcji BP nad komparatorami i placebo, co było powodem przerwania dalszych badań. Ponadto w 4 dużych badaniach antagonistów endoteliny (bozentan, darusentan, enrasentan) u pacjentów z CHF uzyskano sprzeczne wyniki, co tłumaczono wzrostem stężenia endoteliny-1. Dalsze badania antagonistów receptora endoteliny wstrzymano ze względu na działania niepożądane związane z retencją płynów (obrzęki obwodowe, przeciążenia objętościowe). Rozwój tych efektów jest związany z wpływem antagonistów endoteliny na receptory typu B, co zmieniło poszukiwanie leków wpływających na układ endoteliny innymi drogami; i antagoniści receptora endoteliny mają obecnie tylko jedno wskazanie, leczenie nadciśnienia płucnego.
Biorąc pod uwagę duże znaczenie układu endoteliny w regulacji napięcia naczyń, trwają poszukiwania innego mechanizmu działania poprzez wazopeptydazę – EPF, która bierze udział w tworzeniu aktywnej endoteliny-1 (ryc. 3). Blokowanie ACE i połączenie z hamowaniem NEP może skutecznie hamować tworzenie endoteliny-1 i nasilać działanie peptydu natriuretycznego. Zalety podwójnego mechanizmu działania polegają z jednej strony na zapobieganiu wadom inhibitorów NEP związanym z możliwym skurczem naczyń za pośrednictwem aktywacji endoteliny, z drugiej strony działanie natriuretyczne inhibitorów NEP umożliwia kompensację retencji płynów związane z nieselektywną blokadą receptorów endoteliny. Daglutril jest podwójnym inhibitorem NEP i EPF, który znajduje się w fazie II badań klinicznych. Badania wykazały wyraźne kardioprotekcyjne działanie leku ze względu na zmniejszenie przebudowy serca i naczyń, regresję przerostu i zwłóknienia.
Bezpośrednie inhibitory reniny
Wiadomo, że inhibitory ACE i blokery receptora ATII poprzez mechanizm sprzężenia zwrotnego zwiększają aktywność reniny, co jest przyczyną uciekania przed skutecznością blokerów RAAS. Renin stanowi pierwszy krok w kaskadzie RAAS; jest wytwarzany przez komórki przykłębuszkowe nerek. Renina poprzez angiotensynogen sprzyja powstawaniu ATII, zwężaniu naczyń i wydzielaniu aldosteronu, a także reguluje mechanizmy sprzężenia zwrotnego. Dlatego hamowanie reniny umożliwia osiągnięcie pełniejszej blokady układu RAAS. Poszukiwania inhibitorów reniny trwają od lat 70. XX wieku; przez długi czas nie było możliwe uzyskanie doustnej postaci inhibitorów reniny ze względu na ich niską biodostępność w przewodzie pokarmowym (poniżej 2%). Pierwszy bezpośredni inhibitor reniny odpowiedni do podawania doustnego, aliskiren, został zarejestrowany w 2007 roku. Aliskiren ma niską biodostępność (2,6%), długi okres półtrwania (24-40 godzin), pozanerkową drogę eliminacji. Farmakodynamika aliskirenu wiąże się z 80% spadkiem poziomu ATII. W badaniach klinicznych u pacjentów z nadciśnieniem tętniczym aliskiren w dawkach 150-300 mg/dobę prowadził do zmniejszenia SBP o 8,7-13 i 14,1-15,8 mm Hg. odpowiednio, a DBP - o 7,8-10,3 i 10,3-12,3 mm Hg. . Hipotensyjne działanie aliskirenu obserwowano w różnych podgrupach pacjentów, w tym pacjentów z zespołem metabolicznym, otyłością; pod względem nasilenia było ono porównywalne z działaniem inhibitorów ACE, blokerów receptora ATII, a efekt addytywny odnotowano w połączeniu z walsartanem, hydrochlorotiazydem i amlodypiną. Szereg badań klinicznych wykazało działanie ochronne leku na narządy: działanie przeciwbiałkomoczowe u pacjentów z nefropatią cukrzycową (badanie AVOID, n=599), regresja przerostu lewej komory u pacjentów z nadciśnieniem tętniczym (badanie ALLAY, n=465). I tak w badaniu AVOID po 3 miesiącach leczenia losartanem w dawce 100 mg/dobę i osiągnięciu docelowego poziomu ciśnienia tętniczego (<130/80 мм рт.ст.) при компенсированном уровне гликемии (гликированный гемоглобин 8%) больных рандомизировали к приему алискирена в дозах 150-300 мг/сут или плацебо. Отмечено достоверное снижение индекса альбумин/креатинин в моче (первичная конечная точка) на 11% через 3 мес. и на 20% - через 6 мес. в сравнении с группой плацебо. В ночное время экскреция альбумина на фоне приема алискирена снизилась на 18%, а доля пациентов со снижением экскреции альбумина на 50% и более была вдвое большей (24,7% пациентов в группе алискирена против 12,5% в группе плацебо) . Причем нефропротективный эффект алискирена не был связан со снижением АД. Одним из объяснений выявленного нефропротективного эффекта у алискирена авторы считают полученные ранее в экспериментальных исследованиях на моделях диабета данные о способности препарата снижать количество рениновых и прорениновых рецепторов в почках, а также уменьшать профибротические процессы и апоптоз подоцитов, что обеспечивает более выраженный эффект в сравнении с эффектом ингибиторов АПФ . В исследовании ALLAY у пациентов с АГ и увеличением толщины миокарда ЛЖ (более 1,3 см по данным ЭхоКГ) применение алискирена ассоциировалось с одинаковой степенью регресса ИММЛЖ в сравнении с лозартаном и комбинацией алискирена с лозартаном: −5,7±10,6 , −5,4±10,8, −7,9±9,6 г/м2 соответственно. У части пациентов (n=136) проводилось изучение динамики нейрогормонов РААС, и было выявлено достоверное и значительное снижение уровня альдостерона и активности ренина плазмы на фоне применения алискирена или комбинации алискирена с лозартаном, тогда как на фоне применения монотерапии лозартаном эффект влияния на альдостерон отсутствовал, а на активность ренина - был противоположным, что объясняет значимость подавления альдостерона в достижении регресса ГЛЖ.
Ponadto prowadzony jest szereg badań klinicznych aliskirenu w leczeniu innych chorób układu krążenia z oceną wpływu na rokowanie pacjentów: ALOFT (n=320), ASTRONAUT (n=1639), ATMOSPHERE (n =7000) u pacjentów z CHF, badaniu ALTITUDE u pacjentów z cukrzycą i wysokim ryzykiem sercowo-naczyniowym, badaniu ASPIRE u pacjentów z przebudową pozawałową.
Wniosek
Aby rozwiązać problemy zapobiegania chorobom układu krążenia, trwają prace nad tworzeniem nowych leków o złożonym, wielokrotnym mechanizmie działania, które pozwalają na pełniejszą blokadę RAAS poprzez kaskadę mechanizmów regulacji hemodynamicznej i neurohumoralnej. Potencjalne działanie takich leków pozwala nie tylko na uzyskanie dodatkowego efektu hipotensyjnego, ale także na uzyskanie kontroli ciśnienia krwi u pacjentów wysokiego ryzyka, w tym nadciśnienia tętniczego opornego. Leki o wielorakim mechanizmie działania wykazują przewagę w silniejszym działaniu organoprotekcyjnym, co zapobiegnie dalszym uszkodzeniom układu sercowo-naczyniowego. Badanie korzyści ze stosowania nowych leków blokujących RAAS wymaga dalszych badań i oceny ich wpływu na rokowanie pacjentów z nadciśnieniem tętniczym i innymi chorobami układu krążenia.



Literatura
1 Campbell DJ Hamowanie wazopeptydazy: miecz obosieczny? // Nadciśnienie. 2003 Cz. 41. s. 383-389.
2. Laurent S., Schlaich M., Esler M. Nowe leki, procedury i urządzenia na nadciśnienie // Lancet. 2012. Cz. 380. s. 591-600.
3. Corti R., Burnett J.C., Rouleau J.L. i in. Inhibitory wazopeptydazy: nowa koncepcja terapeutyczna w chorobach układu krążenia? // Cyrkulacja. 2001 Cz. 104. s. 1856-1862.
4. Mangiafico S., Costello-Boerrigter LC, Andersen I.A. i in. Hamowanie neutralnej endopeptydazy i układ peptydów natriuretycznych: ewoluująca strategia w terapii sercowo-naczyniowej // Eur. Serce J. 2012, doi:10.1093/eurheartj/ehs262.
5. Rouleau JL, Pfeffer MA, Stewart DJ i in. Porównanie inhibitora wazopeptydazy, omapatrilatu i lizynoprylu na tolerancję wysiłku i zachorowalność u pacjentów z niewydolnością serca: randomizowane badanie IMPRESS // Lancet. 2000 Cz. 356. s. 615-620.
6. Packer M., Califf RM, Konstam M.A. i in. Porównanie omapatrilatu i enalaprylu u pacjentów z przewlekłą niewydolnością serca: Randomizowana próba przydatności Omapatrilat w porównaniu z enalaprylem w ograniczaniu zdarzeń (OVERTURE) // Krążenie. 2002 Cz. 106. s. 920-926.
7. Warner KK, Visconti JA, Tschampel MM Blokery receptora angiotensyny II u pacjentów z obrzękiem naczynioruchowym wywołanym inhibitorem ACE // Ann. Farmakoterapeuta. 2000 Cz. 34. s. 526-528.
8. Kostis J.B., Packer M., Black H.R. i in. Omapatrilat i enalapril u pacjentów z nadciśnieniem tętniczym: badanie Omapatrilat Cardiovascular Treatment vs Enalapril (OCTAVE) // Am. J. Nadciśnienie. 2004 Cz. 17. s. 103-111.
9. Azizi M., Bissery A., Peyrard S. et al. Farmakokinetyka i farmakodynamika inhibitora wazopeptydazy AVE7688 u ludzi // Clin. Farmakol. Ter. 2006 Cz. 79. s. 49-61.
10. Gu J., Noe A., Chandra P. i in. Farmakokinetyka i farmakodynamika LCZ696, nowego podwójnego działania inhibitora neprylizyny receptora angiotensyny (ARNi) // J. Clin. Farmakol. 2010 Cz. 50. s. 401-414.
11. Ruilope LM, Dukat A., Buhm M. et al. Obniżenie ciśnienia krwi za pomocą LCZ696, nowego podwójnego działania inhibitora receptora angiotensyny II i neprylizyny: randomizowane, podwójnie ślepe, kontrolowane placebo, aktywne badanie porównawcze // Lancet. 2010 Cz. 375. s. 1255-1266.
12. Solomon SD, Zile M., Pieske B. et al. Inhibitor neprylizyny receptora angiotensyny LCZ696 w niewydolności serca z zachowaną frakcją wyrzutową: faza 2 podwójnie ślepej próby z randomizacją // Lancet. 2012. Cz. 380(9851). s. 1387-1395.
13. Levin ER Endoteliny // N. Engl. J. Med. 1995 Cz. 333. s. 356-363.
14. Dhaun N., Goddard J., Kohan D.E. i in. Rola endoteliny-1 w nadciśnieniu klinicznym: 20 lat na // Nadciśnienie. 2008 Cz. 52. s. 452-459.
15. Burnier M., Forni V. Antagoniści receptora endoteliny: miejsce w leczeniu samoistnego nadciśnienia tętniczego? // Nefrol. Wybierz. przeszczep. 2011.0:1-4. doi: 10.1093/ndt/gfr704.
16. Krum H., Viskoper RJ, Lacourciere Y. et al. Wpływ antagonisty receptora endoteliny, bozentanu, na ciśnienie krwi u pacjentów z nadciśnieniem pierwotnym. Badacze nadciśnienia bozentanowego // N. Engl. J. Med. 1998 Cz. 338. s. 784-790.
17. Weber MA, Black H., Bakris G. et al. Selektywny antagonista receptora endoteliny w celu obniżenia ciśnienia krwi u pacjentów z nadciśnieniem tętniczym opornym na leczenie: randomizowane, podwójnie ślepe, kontrolowane placebo badanie // Lancet. 2009 Cz. 374. s. 1423-1431.
18. Bakris GL, Lindholm LH, Black H.R. i in. Rozbieżne wyniki przy użyciu klinicznego i ambulatoryjnego ciśnienia krwi: raport z próby nadciśnienia opornego na darusentan // Nadciśnienie. 2010 Cz. 56. s. 824-830.
19. Mann J.F., Green D., Jamerson K. et al. Avosentan na jawną nefropatię cukrzycową // J. Am. towarzyska Nefrol. 2010 Cz. 21. s. 527-535.
20. Kalk P., Sharkovska Y., Kashina E. et al. Enzym konwertujący endotelinę/inhibitor neutralnej endopeptydazy SLV338 zapobiega nadciśnieniowej przebudowie serca w sposób niezależny od ciśnienia krwi // Nadciśnienie. 2011 Cz. 57. s. 755-763.
21. Nussberger J., Wuerzner G., Jensen C. et al. Supresja angiotensyny II u ludzi przez teoretycznie aktywny inhibitor reniny Aliskiren (SPP100): porównanie z enalaprylem // Nadciśnienie. 2002 Cz. 39 ust. 1. P. E1-8.
22. Alreja G., Joseph J. Renin i choroby układu krążenia: przetarta ścieżka czy nowy kierunek? // Świat J. Cardiol. 2011 Cz. 3 ust. 3. s. 72-83.
23. Ingelfinger J.R. Aliskiren i terapia dualna w cukrzycy typu 2 // N. Engl. J. Med. 2008 Cz. 358(23). str. 2503-2505.
24. Pouleur A.C., Uno H., Prescott MF, Desai A. (dla śledczych ALLAY). Supresja aldosteronu pośredniczy w regresji przerostu lewej komory u pacjentów z nadciśnieniem tętniczym // J. Renin-Angiotensin-Aldosterone System. 2011 Cz. 12. s. 483-490.
25. Kelly DJ, Zhang Y., Moe G. i in. Aliskiren, nowy inhibitor reniny, działa renoprotekcyjnie w modelu zaawansowanej nefropatii cukrzycowej u szczurów // Diabetol. 2007 Cz. 50. str. 2398-2404.
Historia badań układu renina-angiotensyna-aldosteron (RAAS), który okazał się najbardziej udany pod względem opracowania podejść do farmakologicznej modulacji jego aktywności, pozwalającej na wydłużenie życia pacjentów z chorobami układu krążenia i nerek , rozpoczął się 110 lat temu. Kiedy zidentyfikowano reninę - pierwszy składnik. Później, w badaniach eksperymentalnych i klinicznych, udało się wyjaśnić fizjologiczną rolę reniny i jej znaczenie w regulacji aktywności RAAS w różnych stanach patologicznych, co stało się podstawą do opracowania wysoce skutecznej strategii terapeutycznej – bezpośrednich inhibitorów reniny.
Obecnie pierwszy bezpośredni inhibitor reniny Rasilez (aliskiren) ma uzasadnienie nawet w sytuacjach, gdy inne blokery RAAS – inhibitory ACE i ARB nie są wskazane lub ich stosowanie jest utrudnione ze względu na rozwój działań niepożądanych.
Kolejną okolicznością, która pozwala liczyć na dodatkowe możliwości bezpośrednich inhibitorów reniny w ochronie narządów docelowych nadciśnienia tętniczego w porównaniu z innymi blokerami RAAS, jest to, że przy stosowaniu leków blokujących RAAS na innych poziomach, zgodnie z prawem ujemnego sprzężenia zwrotnego, dochodzi do jest wzrost stężenia proreniny i wzrost aktywności reninowej osocza. Ta okoliczność niweczy często odnotowywany spadek skuteczności inhibitorów ACE, w tym z punktu widzenia ich zdolności do obniżania podwyższonego ciśnienia krwi. Jeszcze na początku lat 90., kiedy wiele działań ochronnych inhibitorów ACE na narządy nie zostało ustalone tak wiarygodnie jak obecnie, wykazano, że wraz ze wzrostem ich dawki znacząco wzrasta aktywność reninowa osocza i stężenie angiotensyny w osoczu. Wraz z inhibitorami ACE i ARB, diuretyki tiazydowe i pętlowe mogą również powodować zwiększenie aktywności reninowej osocza.
Aliskiren był pierwszym bezpośrednim inhibitorem reniny, którego skuteczność potwierdzono w kontrolowanych badaniach klinicznych III fazy, który ma wystarczający czas działania i obniża podwyższone ciśnienie krwi nawet w monoterapii, a jego przepisywanie można obecnie uznać za nowatorskie podejście do leczenie nadciśnienia. Porównano jego wpływ na stężenie i aktywność poszczególnych składników RAAS w osoczu z inhibitorami ACE i ARB. Okazało się, że aliskiren i enalapryl prawie w równym stopniu zmniejszają stężenie angiotensyny II w osoczu, ale w przeciwieństwie do aliskirenu podawanie enalaprylu prowadziło do ponad 15-krotnego wzrostu aktywności reninowej osocza. Zdolność aliskirenu do zapobiegania negatywnym zmianom w równowadze aktywności składników RAAS wykazano również w porównaniu z ARB.
Zbiorcza analiza badania klinicznego obejmującego łącznie 8481 pacjentów, którzy otrzymywali aliskiren w monoterapii lub placebo, wykazała, że pojedyncza dawka aliskirenu w dawce 150 mg na dobę. lub 300 mg/dzień. spowodowały spadek SBP o 12,5 i 15,2 mm Hg. odpowiednio, w porównaniu z redukcją o 5,9 mmHg, placebo (R<0,0001). Диастолическое АД снижалось на 10,1 и 11,8 мм рт.ст. соответственно (в группе, принимавшей плацебо – на 6,2 мм рт.ст.; Р < 0,0001). Различий в антигипертензивном эффекте алискирена у мужчин и женщин, а также у лиц старше и моложе 65 лет не выявлено.
W 2009 roku opublikowano wyniki wieloośrodkowego kontrolowanego badania klinicznego, w którym porównano skuteczność aliskirenu i hydrochlorotiazydu u 1124 pacjentów z nadciśnieniem tętniczym. W razie potrzeby do tych leków dodawano amlodypinę. Pod koniec okresu monoterapii stało się jasne, że aliskiren prowadzi do wyraźniejszego obniżenia ciśnienia tętniczego niż hydrochlorotiazyd (-17,4/-12,2 mm Hg vs. -14,7/-10,3 mm Hg; R< 0,001)
Bezpośredni inhibitor reniny (aliskiren)
Stymuluje wydzielanie reniny przez nerki, zmniejszając objętość krwi krążącej i perfuzję nerek. Renina z kolei przekształca angiotensynogen w angiotensynę I, prekursor angiotensyny II, która uruchamia kaskadę reakcji prowadzących do wzrostu ciśnienia krwi. Zatem zahamowanie wydzielania reniny może zmniejszyć produkcję angiotensyny II. Podczas przyjmowania diuretyków tiazydowych, inhibitorów ACE i ARB zwiększa się aktywność reninowa osocza. Dlatego hamowanie aktywności reniny może być potencjalnie skuteczną strategią hamowania całego układu renina-angiotensyna. Aliskiren jest pierwszym lekiem nowej klasy – bezpośrednim inhibitorem reniny, dla którego udowodniono działanie hipotensyjne. Poprawiona biodostępność doustnej postaci aliskirenu w porównaniu z dotychczas oferowanymi lekami tego typu oraz długi okres półtrwania pozwalają na przyjmowanie tego leku raz dziennie.
Aliskiren skutecznie obniża ciśnienie krwi zarówno w monoterapii, jak iw połączeniu z diuretykami tiazydowymi (hydrochlorotiazyd), inhibitorami ACE (ramipryl, lizynopryl). ARB (walsartan) lub CCB (amlodypina). Kiedy aliskiren jest przyjmowany z tymi lekami przeciwnadciśnieniowymi, aktywność reniny w osoczu nie zwiększa się, ale pozostaje na poziomie podstawowym lub nawet poniżej niego. Alixiren ma podobne do placebo bezpieczeństwo i tolerancję i nie wchodzi w interakcje z szeroką gamą leków, z wyjątkiem furosemidu. Obecnie dostępne są ograniczone dane dotyczące długoterminowej skuteczności i tolerancji aliskirenu u pacjentów z cukrzycą i nadciśnieniem tętniczym. W rezultacie dokładna rola tego leku w leczeniu nadciśnienia tętniczego u chorych na cukrzycę nie została do końca ustalona.
ALISKIREN (lek Rasilez) - tabletki 150 mg i 300 mg, dawka początkowa 150 mg / 1 raz dziennie, przy niedostatecznej kontroli ciśnienia krwi po 2 tygodniach dawkę można zwiększyć do 300 mg / 1 raz dziennie
Mechanizm akcji. Lek hipotensyjny, selektywny inhibitor reniny o budowie niepeptydowej. Podczas stosowania aliskirenu w monoterapii oraz w skojarzeniu z innymi lekami przeciwnadciśnieniowymi następuje zneutralizowanie tłumienia ujemnego sprzężenia zwrotnego, co skutkuje zmniejszeniem aktywności reninowej osocza (u pacjentów z nadciśnieniem tętniczym średnio o 50-80%), a także stężeniami antytensyny I i II. Po pierwszej dawce nie występuje reakcja hipotensyjna (efekt pierwszej dawki) i odruchowe przyspieszenie akcji serca w odpowiedzi na rozszerzenie naczyń.
Farmakokinetyka. Po podaniu doustnym czas do osiągnięcia maksymalnego stężenia aliskirenu w osoczu wynosi 1-3 godziny, bezwzględna biodostępność wynosi 2,6%. Jednoczesne przyjmowanie pokarmu nie ma istotnego wpływu na farmakodynamikę leku. Dlatego aliskiren można przyjmować z posiłkiem lub bez posiłku. Aliskiren w umiarkowanym stopniu wiąże się z białkami osocza (47-51%), niezależnie od stężenia. Okres półtrwania w fazie eliminacji aliskirenu wynosi 40 godzin (waha się od 34 do 41 godzin). Wydalany jest głównie w postaci niezmienionej przez jelita (91%). Około 1,4% przyjętej dawki jest metabolizowane z udziałem izoenzymu CYP3A4. Po podaniu doustnym około 0,6% aliskirenu jest wydalane przez nerki. Podczas stosowania aliskirenu u pacjentów w wieku powyżej 65 lat nie jest wymagana modyfikacja dawki leku. Farmakokinetyka aliskirenu nie zmienia się istotnie u pacjentów z łagodnymi do umiarkowanych zaburzeniami czynności wątroby (5-9 punktów w skali Childa-Pugha).
interakcje pomiędzy lekami. Prawdopodobieństwo interakcji aliskirenu z innymi lekami jest niskie Podczas stosowania aliskirenu z jednym z następujących leków, jego Cmax lub AUC może ulec zmianie: walsartan (zmniejszenie o 28%), metformina (zmniejszenie o 28%), amlodypina (zmniejszenie o 29%), cymetydyna (wzrost o 19%). Ponieważ w badaniach eksperymentalnych stwierdzono, że P-glikoproteina (błonowy nośnik cząsteczek) odgrywa ważną rolę w regulacji wchłaniania i dystrybucji aliskirenu, możliwa jest zmiana farmakokinetyki tego ostatniego przy jednoczesnym stosowaniu z substancjami hamującymi P -glikoproteina (w zależności od stopnia hamowania). Nie stwierdzono istotnych interakcji aliskirenu ze słabo lub średnio aktywnymi inhibitorami glikoproteiny P, takimi jak atenolol, digoksyna, amlodypina i cymetydyna. Przy równoczesnym stosowaniu z aktywnym inhibitorem atorwastatyny P-glikoproteiny (w dawce 80 mg / dobę) w stanie równowagi obserwuje się wzrost AUC i C max aliskirenu (dawka 300 mg / dobę) o 50%. Przy jednoczesnym podawaniu aktywnego inhibitora glikoproteiny P, ketokonazolu (200 mg) i aliskirenu (300 mg), obserwuje się wzrost Cmax tego ostatniego o 80%. W badaniach eksperymentalnych jednoczesne podawanie aliskirenu z ketokonazolem prowadziło do zwiększenia wchłaniania tego ostatniego z przewodu pokarmowego i zmniejszenia jego wydalania z żółcią. Oczekuje się zmian stężenia aliskirenu w osoczu podczas jednoczesnego stosowania z ketokonazolem lub atorwastatyną w zakresie stężeń określonych przez dwukrotne zwiększenie dawki aliskirenu. W kontrolowanych badaniach klinicznych wykazano bezpieczeństwo aliskirenu w dawce 600 mg i 2-krotnym zwiększeniu maksymalnej zalecanej dawki terapeutycznej. Podczas jednoczesnego stosowania aliskirenu z ketokonazolem lub atorwastatyną nie jest wymagana modyfikacja dawki aliskirenu. W przypadku stosowania z tak wysoce aktywnym inhibitorem glikoproteiny P, jak cyklosporyna (200 i 600 mg), zdrowe osoby wykazały wzrost C max i AUC aliskirenu (75 mg) odpowiednio o 2,5 i 5 razy (nie zaleca się stosowania aliskiren jednocześnie z cyklosporyną). Przy równoczesnym stosowaniu aliskirenu z furosemidem następuje zmniejszenie AUC i Cmax furosemidu odpowiednio o 28% i 49%. Aby zapobiec możliwemu zatrzymaniu płynów podczas przepisywania aliskirenu razem z furosemidem na początku iw trakcie leczenia, konieczne jest dostosowanie dawki furosemidu w zależności od efektu klinicznego. Należy zachować ostrożność podczas jednoczesnego stosowania aliskirenu z solami potasu, lekami moczopędnymi oszczędzającymi potas, substytutami soli kuchennej zawierającymi potas lub innymi produktami leczniczymi, które mogą zwiększać stężenie potasu we krwi.
Efekt uboczny. Z układu pokarmowego: często - biegunka. Reakcje skórne: czasami - wysypka skórna Od strony parametrów laboratoryjnych: rzadko - nieznaczne obniżenie stężenia hemoglobiny i hematokrytu (średnio odpowiednio o 0,05 mmol/l i 0,16%), które nie wymagało przerwania leczenia, niewielki wzrost stężenia potasu w surowicy krwi (0,9% w porównaniu do 0,6% w przypadku placebo). Reakcje alergiczne: w niektórych przypadkach - obrzęk naczynioruchowy.
Przeciwwskazania i ograniczenia. Przeciwwskazania: dzieci i młodzież do 18 roku życia, ciąża, laktacja (karmienie piersią), nadwrażliwość na aliskiren. Stosowanie w okresie ciąży i laktacji (karmienie piersią) jest przeciwwskazane.
Skuteczność i bezpieczeństwo stosowania aliskirenu u pacjentów z ciężkimi zaburzeniami czynności wątroby (powyżej 9 punktów w skali Childa-Pugha) nie zostało ustalone.
Skuteczność i bezpieczeństwo stosowania aliskirenu nie zostały ustalone: u pacjentów z ciężkimi zaburzeniami czynności nerek (stężenie kreatyniny w surowicy >150 µmol/l u kobiet i >177 µmol/l u mężczyzn i (lub) wskaźnik przesączania kłębuszkowego mniejszy niż 30 ml/min), z zespołem nerczycowym, nadciśnieniem nerkowo-naczyniowym oraz podczas regularnej hemodializy.
Ostrożnie alisikiren należy stosować u pacjentów z jednostronnym lub obustronnym zwężeniem tętnicy nerkowej lub zwężeniem tętnicy pojedynczej nerki, cukrzycą, zmniejszonym BCC, hiponatremią, hiperkaliemią lub po przeszczepieniu nerki.
Skuteczność i bezpieczeństwo stosowania aliskirenu nie zostały ustalone: u pacjentów z ciężkimi zaburzeniami czynności nerek (stężenie kreatyniny w surowicy >150 µmol/l u kobiet i >177 µmol/l u mężczyzn i (lub) wskaźnik przesączania kłębuszkowego mniejszy niż 30 ml/min), z zespołem nerczycowym, nadciśnieniem nerkowo-naczyniowym oraz w trakcie regularnej hemodializy, a także u pacjentów z ciężkimi zaburzeniami czynności wątroby (powyżej 9 punktów w skali Childa-Pugha), u pacjentów z jednostronnym lub obustronnym zwężeniem tętnic nerkowych lub zwężeniem tętnicy nerkowej tętnica pojedynczej nerki.
U pacjentów z cukrzycą podczas terapii aliskirenem w skojarzeniu z inhibitorem ACE obserwowano zwiększenie częstości występowania hiperkaliemii (5,5%). Podczas stosowania aliskirenu i innych leków wpływających na RAAS u pacjentów z cukrzycą konieczne jest regularne monitorowanie składu elektrolitów w osoczu krwi i czynności nerek.
Na tle terapii aliskirenem możliwy jest wzrost stężenia potasu, kreatyniny i azotu mocznikowego we krwi, co jest charakterystyczne dla leków wpływających na RAAS. Na początku leczenia aliskirenem u pacjentów ze zmniejszonym BCC i (lub) hiponatremią (w tym na tle dużych dawek leków moczopędnych) możliwe jest objawowe niedociśnienie tętnicze. Przed użyciem należy przeprowadzić korektę naruszeń równowagi wodno-solnej. U pacjentów ze zmniejszonym BCC i/lub hiponatremią leczenie należy prowadzić pod ścisłą kontrolą lekarską.
Układ renina-angiotensyna-aldosteron odgrywa kluczową rolę w regulacji ciśnienia krwi oraz gospodarki wodno-elektrolitowej. Bezpośredni inhibitor reniny - aliskiren, poprzez zmniejszenie aktywności reniny osocza, działa kardio- i nefroprotekcyjnie. Działanie przeciwnadciśnieniowe nie zależy od płci, rasy, wieku, wskaźnika masy ciała. Działanie przeciwnadciśnieniowe aliskirenu i inhibitorów konwertazy angiotensyny, blokerów receptora angiotensyny II i antagonistów wapnia jest porównywalne. Aliskiren jest skuteczny u pacjentów z otyłością, cukrzycą, zaburzeniami czynności nerek i zespołem metabolicznym.
Bezpośrednie inhibitory reniny – aliskiren w leczeniu nadciśnienia tętniczego
Układ renina-angiotensyna-aldosteron odgrywa kluczową rolę w regulacji ciśnienia krwi oraz gospodarki wodno-elektrolitowej. Bezpośredni inhibitor reniny - aliskiren, zmniejszający aktywność reninową osocza, wykazujący działanie kardioprotekcyjne i nefroprotekcyjne. Działanie przeciwnadciśnieniowe jest niezależne od płci, rasy, wieku, wskaźnika masy ciała. Działanie przeciwnadciśnieniowe aliskirenu i inhibitorów konwertazy angiotensyny, blokerów receptora angiotensyny II, antagonistów wapnia jest porównywalne. Aliskiren jest skuteczny u pacjentów z otyłością, cukrzycą, zaburzeniami czynności nerek i zespołem metabolicznym.
W trakcie badania układu renina-angiotensyna-aldosteron (RAAS) opracowano metody regulacji jego aktywności farmakologicznej. Pierwszy składnik RAAS, renina, został zidentyfikowany 110 lat temu. Później wykazano jego znaczenie w regulacji aktywności RAAS w stanach patologicznych, co stało się podstawą do opracowania bezpośrednich inhibitorów reniny (DRI). RAAS odgrywa kluczową rolę w regulacji ciśnienia krwi (BP) oraz równowagi wodno-elektrolitowej. Wzrost aktywności RAAS odgrywa ważną rolę w powstawaniu i progresji nadciśnienia tętniczego (AH), przewlekłej niewydolności serca (CHF), przewlekłej choroby nerek i miażdżycy układowej. RAAS bierze bezpośredni udział w procesach wzrostu i różnicowania tkanek, modulacji stanu zapalnego i apoptozy, a także nasilenia syntezy i wydzielania szeregu substancji neurohumoralnych. Główne efekty RAAS realizowane są poprzez angiotensynę II (ATII) poprzez stymulację specyficznych receptorów. Aktywacja receptora angiotensyny podtypu 1 (AT1) prowadzi do skurczu naczyń, stymuluje uwalnianie wazopresyny, aldosteronu, endoteliny, noradrenaliny. Fizjologiczna rola innych podtypów receptora angiotensyny (AT3, AT4 i ATx) jest nadal badana. ATII przyczynia się do gromadzenia macierzy kolagenowej, produkcji cytokin, cząsteczek adhezyjnych, aktywacji systemu sygnalizacji wewnątrzkomórkowej, zwiększonej ekspresji genów fenotypowych płodu, odgrywa ważną rolę w przebudowie mięśnia sercowego i przeroście lewej komory (LV), ATII uczestniczy w procesach przebudowy tętnic, nasilenia stresu oksydacyjnego i apoptozy, przyczynia się do powstawania i progresji nadciśnienia tętniczego, CHF, miażdżycowego uszkodzenia naczyń, nefropatii cukrzycowej i niecukrzycowej, angiopatii w cukrzycy (DM), rzucawki ciężarnych, choroby Alzheimera. Postęp chorób sercowo-naczyniowych nie zależy od wazopresyjnego działania ATII.
Wydzielanie reniny jest pierwszym etapem zwiększania syntezy ATI, ATII i innych produktów kaskady RAAS. Realizacja kolejnych efektów RAAS jest modulowana przez wpływ reniny na określone receptory, indukując wzrost ATII.
Do niedawna istniały następujące inhibitory RAAS – inhibitory konwertazy angiotensyny (inhibitory ACE) i blokery receptora ATII (ARB). Mechanizm działania inhibitorów ACE jest następujący: aktywność ACE jest hamowana, co prowadzi do zmniejszenia działania ATII i spowolnienia degradacji wazopresorów (bradykininy i prostaglandyny E 2). ARB kompetycyjnie hamują receptory ATII i zmniejszają działanie ATII. Receptory dla reniny i proreniny znajdują się na powierzchni komórki. Aktywacja komórkowego szlaku sygnałowego przez reninę prowadzi do zwłóknienia i hipertrofii komórkowej. W ostatnich latach aktywność RAAS kontrolowano poprzez ograniczenie produkcji ATII, blokadę receptorów ATII i aldosteronu, w wyniku ograniczenia wydzielania reniny, głównie poprzez stosowanie β-adrenolityków. Liczne badania wykazały, że odpowiednie obniżenie aktywności RAAS przy pomocy inhibitorów ACE, ARB czy aldosteronu jest raczej postulowane niż faktycznie osiągane, gdyż zjawisko „ucieczki” przeciwnadciśnieniowego i narządowo ochronnego działania blokerów RAAS rozwija się w trakcie ich długotrwałego stosowania. używać. Aby przezwyciężyć to zjawisko stosuje się kombinacje inhibitor ACE + ARB, inhibitor ACE + β-bloker, inhibitor ACE + spironolakton. Pojawienie się PIR jest postrzegane jako sposób na osiągnięcie pełniejszej kontroli nad aktywnością RAAS i przezwyciężenie zjawiska „ucieczki”.
Pierwsze PIR zostały zsyntetyzowane w latach 70. XX wieku, ale pierwszym lekiem odpowiednim do podawania doustnego był aliskiren (A). A., wiążąc się z aktywną częścią cząsteczki docelowej, zapobiega jej interakcji z angiotensynogenem. Zmniejszając aktywność reniny osocza (ARP), A. wykazuje działanie kardioprotekcyjne i nefroprotekcyjne. Inhibitory RAAS stymulują ARP, powodując następujące efekty: zwężenie naczyń w kłębuszkach nerkowych, zapalenie, zwłóknienie (nerki); przerost, zwłóknienie, zwężenie naczyń (serce); rozrost, przerost, stan zapalny, utlenianie lipidów, zwłóknienie (naczyń); zwężenie naczyń (mózg). A. działa w miejscu aktywacji RAAS i zmniejsza ARP. W przeciwieństwie do inhibitorów ACE i ARB, A zmniejsza poziomy ATI, AII i ARP. Renina ma aktywność enzymatyczną, jak również za pośrednictwem receptorów.
Farmakokinetyka A. Badania kliniczne wykazały tolerancję porównywalną z placebo. Czas działania tego leku przekracza 24 godziny, a rozszerzenie naczyń nerkowych może utrzymywać się do 48 godzin. Okres półtrwania A wynosi około 40 godzin, co zapewnia pojedynczą dawkę dziennie. Zalecana dawka początkowa A wynosi 150 mg z dalszym zwiększeniem do 300 mg. Właściwości farmakokinetyczne A nie zależą od glikemii na czczo i stężenia hemoglobiny glikozylowanej w osoczu. Eliminacja leku odbywa się w postaci niezmienionej z żółcią, wydalanie z moczem<1%. Исследования первой и второй фазы показали, что препарат способствует эффективной блокаде РААС и дозозависимому предотвращению повышения уровня АД . Полный антигипертензивный эффект наступает через 2 недели и не зависит от пола, расы, возраста, индекса массы тела. А обладает минимальным риском лекарственных взаимодействий, не требует коррекции дозы при хронической почечной недостаточности (ХПН), при поражении печени. Добавление А к ловастатину, атенололу, варфарину, фуросемиду, дигоксину, целекоксибу, гидрохлоротиазиду (ГХТЗ), рамиприлу, валсартану, метформину и амлодипину не приводило к клинически значимому увеличению экспозиции А. Совместное его применение с аторвастатином приводило к 50% увеличению Cmax (максимальная концентрацию препарата) и AUC (площадь под кривой «концентрация - время») после приема нескольких доз. Совместное применение 200 мг кетоконазола 2 раза в день с А приводило к 80% увеличению уровня А в плазме. При совместном применении А с фуросемидом AUC и Cmax фуросемида снижались на 30 и 50% соответственно . Не требуется коррекции дозы А у пациентов с ХПН. У пациентов с ХПН отмечается умеренное (~двухкратное) увеличение экспозиции А, но оно не коррелировало с тяжестью поражения почек и клиренсом креатинина. Клиренс А составлял 60–70 % у здоровых. Почечный клиренс А уменьшался с увеличением тяжести поражения почек. Поскольку поражение почек оказывает только умеренное влияние на экспозицию А, то коррекция дозы А, скорее всего, не требуется у пациентов с гипертензией и поражением почек. Не требуется коррекции дозы А и у пациентов поражением печени. Не было отмечено достоверной корреляции между экспозицией А и тяжестью поражения печени. А способен осуществлять блокаду РААС, что приводит к снижению сосудистого тонуса и системного АД. Однако препарат не лишен и негативных качеств, связанных с феноменом «ускользания», что характерно для всех лекарственных средств, блокирующих активность РААС. Снижение эффективности А вследствие восстановления секреции ренина или наличия синдрома отмены не подтверждается клиническими наблюдениями .
Skuteczność hipotensyjna A. ARP jest wskaźnikiem niezbędnym nie tylko w diagnostyce rzadkich wtórnych postaci nadciśnienia tętniczego (nerkowo-naczyniowego). Kliniczne i prognostyczne znaczenie ARP jest następujące: wskaźnik wzrasta wraz z nadciśnieniem w połączeniu z innymi czynnikami ryzyka (płeć męska, palenie tytoniu, cukrzyca typu 2, otyłość (Ob.), zespół metaboliczny) oraz w przypadku uszkodzenia narządu docelowego ( TOM) (utrzymujący się spadek współczynnika przesączania kłębuszkowego); wzrost reniny ARP może być jatrogenny, prowokowany przez inhibitory ACE i/lub diuretyki (pętlowe, tiazydowe), powodując nerkową utratę sodu: obserwuje się dalszą aktywację RAAS, co prowadzi do utraty kontroli nad ciśnieniem tętniczym i progresji CHF; wzrost ARP zawsze predysponuje do zaostrzenia POM i potencjalnie śmiertelnych powikłań sercowo-naczyniowych i nerkowych; podwyższony poziom ARP jest niezależnym czynnikiem działania farmakologicznego PIR, co umożliwia uzyskanie obniżenia ciśnienia krwi i zahamowanie progresji POM. A. może pretendować do roli skutecznego leku hipotensyjnego w monoterapii iw skojarzeniu z innymi lekami. Wskazaniami do zastosowania PIR są: hiperreninowe odmiany nadciśnienia tętniczego, nadciśnienie normoreninowe, w którym prorenina i pośredniczona aktywacja receptorów proreninowych prowadzi do destrukcji tkanek. PIR jest wskazana nie tylko w nadciśnieniu nerkowo-naczyniowym i CHF, ale także przy zwiększonym stężeniu proreniny w osoczu (nadciśnienie z nadmierną aktywacją współczulnego układu nerwowego, zespół metaboliczny, cukrzyca typu 2, menopauza).
Monoterapia A. zapewnia zależne od dawki obniżenie ciśnienia rozkurczowego (DBP) i skurczowego (SBP) u pacjentów z łagodnym do umiarkowanego nadciśnieniem tętniczym. Ocena skuteczności i bezpieczeństwa A. u 672 pacjentów z nadciśnieniem tętniczym I-II stopnia (st.) w 8-tygodniowym badaniu kontrolowanym placebo wykazała zależne od dawki zmniejszenie SBP i DBP. Przeciwnadciśnieniowe działanie A utrzymywało się przez dwa tygodnie po jego odstawieniu; A był dobrze tolerowany; częstość działań niepożądanych nie różniła się od placebo. A - nazwa handlowa rasilez (P) - w dawce 150 mg obniża SBP o 13 mm Hg. Art. i DBP o 10,3 mm Hg. Art., aw dawce 300 mg obniża SBP z 15 do 22 mm Hg. Sztuka. (w zależności od art. AG) i DBP - o 11 mm Hg. Sztuka. A zapewnia kontrolę ciśnienia krwi w okresie wczesnoporannym. Po anulowaniu A nie występuje zjawisko „odbicia”. Zbiorcza analiza badań klinicznych obejmujących 8481 pacjentów. leczonych A w monoterapii lub placebo wykazali, że pojedyncza dawka A w dawce 150 lub 300 mg/dobę spowodowała spadek SBP o 12,5 i 15,2 mm Hg. Sztuka. odpowiednio, w porównaniu ze spadkiem o 5,9 mm Hg. Sztuka. u pacjentów otrzymujących placebo (str<0,0001). ДАД снижалось на 10,1 (на дозе 150 мг) и 11,8 мм рт. ст. (на дозе 300 мг) соответственно (в группе плацебо - на 6,2 мм рт. ст., р<0,0001). Различий в антигипертензивном эффекте А у мужчин и женщин, а также у лиц старше и моложе 65 лет не выявлено. При применении иАПФ увеличиваются концентрации проренина и АРП (снижается эффективность иАПФ). При увеличении дозы иАПФ достоверно нарастает АРП и плазменная концентрация АТI . Исследование А в сравнении с иАПФ у пациентов с мягкой и умеренной АГ установило следующее: А достоверно больше снижает ДАД и САД, чем рамиприл через 12 недель лечения (монотерапия). А ± гидрохлортиазид (ГХТЗ) достоверно больше снижает ДАД и САД, чем рамиприл ± ГХТЗ через 26 недель лечения. А достоверно больше снижает ДАД и САД, чем рамиприл через 12 недель лечения (монотерапия) у пациентов с АГ II ст. Терапия А обеспечивает достоверно лучший контроль АД по сравнению с рамиприлом. САД и ДАД возвращаются к исходному уровню более быстро после отмены рамиприла, чем после отмены А. Сравнение гипотензивной эффективности А, ирбесартана и рамиприла после пропущенной дозы показало следующее: после пропущенной дозы достигнутое снижение АД было достоверно больше в группе А., чем в группе рамиприла . Достоверно больший процент снижения АД поддерживается после пропущенной дозы А по сравнению с ирбесартаном или рамиприлом. Возвращение к исходному АД происходит более плавно после отмены А., чем рамиприла. А. и эналаприл почти в равной степени уменьшают плазменную концентрацию АТП, но в отличие от А прием эналаприла приводил к более чем 15-кратному росту АРП. В условиях низкосолевой диеты индуцированная А органная (в частности, почечная) вазодилатация может сохраняться до 48 часов. Провоцировать подъем АРП могут препараты, стимулирующие натрийурез (тиазидовые и петлевые диуретики). Назначение А. в этой ситуации один из наиболее действенных подходов к устранению реактивного повышения АРП при комбинации с иАПФ и тиазидовым диуретиком.
W 2009 roku opublikowano wyniki wieloośrodkowego kontrolowanego badania klinicznego, w którym porównano skuteczność A i HCTZ (początkowa terapia hipotensyjna) u 1124 pacjentów z nadciśnieniem tętniczym; w razie potrzeby do tych leków dodawano amlodypinę. Pod koniec okresu monoterapii (tydzień 12) stało się jasne, że A prowadzi do wyraźniejszego obniżenia ciśnienia krwi niż HCTZ (-17,4/-12,2 mm Hg vs. -14,7/-10,3 mm Hg Hg, r<0,001). Эти результаты важны, поскольку большинство пациентов, страдающих АГ, исходно нуждаются в комбинированной антигипертензивной терапии. Оптимизация комбинированной антигипертензивной терапии важна у пациентов с Ож. при этом у А имеются дополнительные преимущества . Больные с Ож, у которых полная (25 мг/сут) доза ГХТЗ не приводила к снижению АД, были рандомизированы на группы, которым назначали амлодипин + ГХТЗ (10/25 мг/сут), ирбесартан + ГХТЗ (300/25 мг/сут) и А. + ГХТЗ (300/25 мг/сут). По мере нарастания ст. Ож антигипертензивная эффективность схем лечения (БРА + ГХТЗ, антагонист кальция + ГХТЗ) снижается. В группе с Ож. III ст. (ИМТ≥40 кг/м 2) только у 50% удалось достичь целевого АД с помощью ирбесартана + ГХТЗ, у 43,8% - с помощью амлодипина + ГХТЗ и лишь у 16,7% - с помощью ГХТЗ. При менее выраженном (I–II ст.) Ож более чем у 40% пациентов, получавших БРА + ГХТЗ или амлодипин + ГХТЗ, и более чем у 60% больных, принимавших только ГХТЗ, целевое АД не было достигнуто. В группе пациентов с Ож. I-II ст., получавших А. + ГХТЗ, целевого АД достигли 56,7% больных, а с Ож. III ст. - 68,8%. АГ, сочетающаяся с Ож, часто ассоциируется с увеличением активности РААС и трудно корригируется, поэтому в таких случаях может быть назначен А.
Ustalono zdolność A. do obniżania ciśnienia krwi i zmniejszania albuminurii. W badaniu AVOID z udziałem 599 pacjentów z nefropatią cukrzycową i nadciśnieniem oceniano wpływ połączenia maksymalnych dawek losartanu i A na albuminurię (na podstawie stosunku albumina/kreatynina w moczu). Dodatkowi A (300 mg/dobę) do losartanu (100 mg/dobę) towarzyszyło istotne obniżenie stosunku albumina/kreatynina w moczu o 20% w całej grupie (100%), a o 24,7% przez 50% lub więcej. W grupie otrzymującej losartan + placebo zmniejszenie stosunku albuminy do kreatyniny w moczu o 50% lub więcej osiągnięto tylko u 12,5% pacjentów (p<0,001). ПИР могут уменьшить альбуминурию как в режиме монотерапии, так и при комбинации с БРА, позволяющей достичь оптимальной ст. блокады РААС, обеспечивающей устранение генерализованной и локально-почечной дисфункции эндотелия.
I ze skojarzoną terapią nadciśnienia. U pacjentów z łagodnym do umiarkowanego nadciśnieniem bez i z A. + HCTZ zapewniają znaczną redukcję DBP i SBP. Więcej pacjentów osiąga kontrolę BP przy kombinacji A + HCTZ niż po innych kombinacjach HCTZ. U pacjentów z nadciśnieniem tętniczym i cukrzycą A + ramipryl istotnie lepiej obniża ciśnienie krwi niż oba składniki monoterapii. A zapewnia znacznie lepszą kontrolę ciśnienia krwi niż ramipryl. U pacjentów z łagodnym do umiarkowanego nadciśnieniem tętniczym A + walsartan znacznie lepiej obniża ciśnienie krwi niż oba składniki monoterapii. A znacząco obniża ciśnienie krwi w połączeniu z amlodypiną w dawce 5 mg/dobę. A zwiększa poziom kontroli ciśnienia krwi w porównaniu z amlodypiną w dawce 5 mg/dobę. A. ± HCTZ są skuteczne w długotrwałej terapii nadciśnienia tętniczego. A + walsartan ± HCTZ zapewniają długoterminową skuteczność hipotensyjną (analiza pośrednia 6-miesięcznej terapii).
W 2009 roku opublikowano projekt badania ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints) (część programu ASPIRE HIGHER), w którym bada się wpływ podwójnej blokady RAAS przy użyciu kombinacji A i standardowej terapii (inhibitor ACE lub ARB) u pacjentów z cukrzycą typu 2 z dużym ryzykiem powikłań sercowo-naczyniowych i nerkowych, częściowo z powodu wzrostu RDA. Głównym celem tego badania jest ocena skuteczności dodania A do standardowej terapii na wpływ na złożony punkt końcowy (zgon z przyczyn sercowo-naczyniowych i powikłania: udana resuscytacja, zawał serca niezakończony zgonem, udar mózgu niezakończony zgonem, nieplanowana hospitalizacja z powodu CHF; rozwój schyłkowej niewydolności nerek, podwojenie stężenia kreatyniny w surowicy, zgon z przyczyn związanych z uszkodzeniem nerek). Badanie to powinno trwać około 4 lat, a jego wyniki mają uzasadniać zastosowanie kombinacji A. z inhibitorem ACE lub ARB w celu zahamowania progresji zespołu sercowo-nerkowego w cukrzycy typu 2. Największej skuteczności A można spodziewać się w tych odmianach nadciśnienia tętniczego, w których występuje tendencja do wzrostu ARP (występujące nadciśnienie samoistne, obj., zespół metaboliczny, cukrzyca typu 2, przewlekła niewydolność nerek). Zdecydowana większość pacjentów z nadciśnieniem tętniczym już w pierwszym etapie leczenia wymaga skojarzonej terapii hipotensyjnej i jak wykazano w jednym z niedawno opublikowanych badań klinicznych, w ramach skojarzeń A zachowuje swoją aktywność niezależnie od początkowego ARP. Wzrost ARP u pacjentów z nadciśnieniem tętniczym jest uważany za marker diagnostyczny i niezależny czynnik ryzyka potencjalnie śmiertelnych incydentów sercowo-naczyniowych. Farmakologiczna modulacja ARP jest jednym z najbardziej obiecujących podejść do zarządzania ryzykiem CVD u pacjentów z AH związaną z uszkodzeniem nerek, zespołem metabolicznym i otyłością. . Badanie AVOID (Aliskiren in the eValuation of proteinuria In Diabetes) (część programu ASPIRE HIGHER) ma na celu ocenę potencjału danego leku hipotensyjnego w zakresie ochrony narządów docelowych w różnych sytuacjach charakteryzujących się bardzo dużym ryzykiem potencjalnie śmiertelnego powikłania (przerost LV, DM typu 2). , HSN). Tymczasowe wyniki sugerują, że bezpośrednia blokada A reniny jest jedną z najbardziej dostępnych strategii poprawy długoterminowego rokowania. W badaniu ALLAY (The Aliskiren Left Ventricular Assessment of Hypertrophy) A spowodował zmniejszenie wskaźnika masy mięśnia lewej komory, odzwierciedlające regresję jego przerostu, u pacjentów z nadciśnieniem tętniczym i nadwagą. Połączenie A i losartanu spowodowało dalsze zmniejszenie wskaźnika masy mięśnia lewej komory o dodatkowe 20% w porównaniu z samym losartanem, jednak różnica ta nie osiągnęła wartości istotnej statystycznie. Zgodnie z wynikami badania ALOFT (ALiskiren Observation of heart Failure Treatment study) dodanie A do standardowego schematu leczenia CHF z objawami złego rokowania (utrzymujący się wzrost stężenia peptydu natriuretycznego w osoczu) i nadciśnieniem jeszcze bardziej poprawiło stosunek od wielkości niedomykalności zastawki mitralnej do okolicy ujścia zastawki mitralnej i transmisyjnego przepływu krwi. Dzięki A udało się uzyskać obniżenie stężenia markerów nieadaptacyjnej aktywacji neurohumoralnej (poziomy w osoczu mózgowego peptydu natriuretycznego i jego N-aminoterminalnego prekursora (NT-pro BNP), stężenie aldosteronu w moczu, ARP). Perspektywy zastosowania A w celu zahamowania rozwoju uszkodzenia nerek determinuje jego wysokie bezpieczeństwo, oczywiście znacząco przewyższające inne blokery RAAS (inhibitory ACE, ARB i antagoniści aldosteronu) ze względu na mniejsze ryzyko wzrostu kreatyninemii i potasu. Wydalany głównie z żółcią, a nie z moczem, A zachowuje swoje działanie przeciwnadciśnieniowe, ale nie upośledza czynności nerek u pacjentów z utrzymującym się spadkiem szybkości przesączania kłębuszkowego. To właśnie w nefrologii agresywna blokada RAAS za pomocą kilku klas leków stosowanych jednocześnie może być skuteczna w zapobieganiu schyłkowej niewydolności nerek. A zmniejsza albuminurię (znacznie przewyższającą monoterapię każdym z leków) oraz prawdopodobieństwo nieodwracalnego pogorszenia funkcji nerek w grupie pacjentów (z białkomoczem > 1 g/dobę), co wykazano w badaniu COOPERATE (Combination treatment of angiOtensin -II bloker receptora Er i inhibitor enzymu konwertującego angiotensynę w niecukrzycowej chorobie nerek). Badanie ONTARGET (OngoiNg Telmisartan Alone and in connection with Ramipril Global Endpoint Trial) wykazało, że połączenie inhibitorów ACE i ARB wiąże się z większym prawdopodobieństwem wystąpienia niedociśnienia tętniczego i hiperkaliemii oraz, w porównaniu z monoterapią tymi lekami, wiąże się z zwiększenie częstości rozpoczynania programu hemodializy i podwojenie kreatyninemii u chorych z grupy wysokiego ryzyka z POM. Obiektem wykazania właściwości nefroprotekcyjnych A. mogą być następujące sytuacje kliniczne: nadciśnienie tętnicze/zespół metaboliczny lub cukrzyca typu 2 z albuminurią; AH związana z trwałym spadkiem przesączania kłębuszkowego; Nadciśnienie w przewlekłej chorobie nerek z białkomoczem (w tym nerczycowym) i bez niego (na przykład nefropatia cewkowo-śródmiąższowa); nadciśnienie naczyniowo-nerkowe różnego pochodzenia; pacjenci, u których z różnych przyczyn występuje wzrost kreatyninemii lub hiperkaliemii podczas stosowania inhibitorów ACE lub ARB; śmiertelna przewlekła niewydolność nerek, w tym leczonych programową hemodializą lub stałą ambulatoryjną dializą otrzewnową; biorcy przeszczepu nerki.
Nowa klasa leków przeciwnadciśnieniowych (AID) wymaga dalszych badań, aby zwiększyć ilość dowodów na spowolnienie postępu POM.
A, oczywiście, jest wskazany dla większości kategorii pacjentów cierpiących na nadciśnienie, a okoliczność ta znajduje odzwierciedlenie w rosyjskich zaleceniach dotyczących nadciśnienia w diagnostyce i leczeniu nadciśnienia tętniczego (czwarta wersja, 2010) jako dodatkowej klasy leków przeciwnadciśnieniowych do terapii skojarzonej. Wraz z rozwojem uszkodzenia nerek A może skutecznie zapobiegać schyłkowej niewydolności nerek i poprawiać rokowanie u tych pacjentów.
NA. Andriejczew, Z.M. Galejewa
Kazański Państwowy Uniwersytet Medyczny
Andreichev Nail Alexandrovich - Kandydat nauk medycznych, profesor nadzwyczajny, Katedra Terapii Wydziału i Kardiologii
Literatura:
1. Bauer J.H., Reams GP. Antagoniści receptora angiotensyny II typu 1. Nowa klasa leków hipotensyjnych // Arch. Stażysta. Med. - 1995. - Cz. 155 (13). - R. 1361-1368.
2. Kim S., Iwao H. Mechanizmy molekularne i komórkowe chorób sercowo-naczyniowych i nerek, w których pośredniczy angiotensyna II // Pharmacol. Obrót silnika. - 2000. - Cz. 52 ust. 1. - R. 11-34.
3. Brązowy M. J. Aliskiren // Cyrkulacja. - 2008. - Cz. 118(7). - R. 773-784.
4. Nguyen G., Delarue F., Burckle C. et al. Kluczowa rola receptora renina/prorenina w produkcji angiotensyny II i odpowiedzi komórkowej na reninę // Clin. Inwestować. - 2002. - Cz. 109(11). - R. 1417-1427.
5. Nussberger J., Wuerzner G., Jensen C. et al. Supresja angiotensyny II u ludzi przez doustny aktywny inhibitor reniny Aliskiren (SPP100): porównanie z enalaprylem // Nadciśnienie. - 2002. - Cz. 39 ust. 1. - R. 1-8.
6. Zhao C., Vaidyanathan S., Dieterich HA i in. Ocena interakcji farmakokinetycznych między doustnym bezpośrednim inhibitorem reniny aliskirenem i furosemidem: badanie na zdrowych ochotnikach // Clin. Farmakol. Ter. - 2007. - Cz. 81 (Dodatek 1). - S. 110 (PIII-78).
7. Gradman A.H., Kad R. Hamowanie reniny w nadciśnieniu tętniczym // Am. kol. kardiol. - 2008. - Cz. 51(5). - S. 519-528.
8. Oh B.-H., Mitchell J., Herron J.R. i in. Aliskiren, doustny inhibitor reniny, zapewnia zależną od dawki skuteczność i stałą 24-godzinną kontrolę ciśnienia krwi u pacjentów z nadciśnieniem tętniczym // Am. kol. kardiol. - 2007. - Cz. 49. - S. 1157-1163.
9. Fisher ND, Jan Danser AH, Nussberger J. et al. Nerkowe i hormonalne odpowiedzi na bezpośrednie hamowanie reniny za pomocą aliskirenu u zdrowego człowieka // Krążenie. - 2008. - Cz. 117 (25). - S. 3199-3205.
10. Dahlo FB, Anderson DR, Arora V. et al. Aliskiren, bezpośredni inhibitor reniny, zapewnia skuteczność hipotensyjną i doskonałą tolerancję niezależnie od wieku i płci u pacjentów z nadciśnieniem tętniczym (abstr) // Clin. nadciśnienie. - 2007. - Cz. 9 (Dodatek A). - SA157.
11. Andersen K., Weinberger MH, Egan B. et al. Porównawcza skuteczność i bezpieczeństwo aliskirenu, doustnego bezpośredniego inhibitora reniny i ramiprylu w nadciśnieniu: 6-miesięczna, randomizowana, podwójnie ślepa próba // Nadciśnienie. - 2008. - Cz. 26. - S. 589-599.
12. Schmieder RE, Philipp T., Guerediaga J. et al. Długotrwała skuteczność przeciwnadciśnieniowa i bezpieczeństwo doustnego bezpośredniego inhibitora reniny Aliskiren. 12-miesięczna randomizowana, podwójnie ślepa próba porównawcza z hydrochlorotiazydem // krążeniem. - 2009. - Cz. 119. - S. 417-425.
13. Prescott MF, Boye SW, Le Breton S. et al. Skuteczność przeciwnadciśnieniowa bezpośredniego inhibitora reniny aliskirenu dodanego do leczenia hydrochlorotiazydem u pacjentów ze skrajną otyłością i nadciśnieniem tętniczym // Am. kol. kardiol. - 2007. - Cz. 49 (9, dodatek A). - S. 370A. - (str. 1014-169).
14. Persson F., Rossing P., Schjoedl K.J. i in. Przebieg czasowy przeciwbiałkowego i przeciwnadciśnieniowego działania bezpośredniego hamowania reniny w cukrzycy typu 2 // Kidney Int. - 2008. - Cz. 73(12). - S. 1419-1425.
15. Parving H.H., Persson F., Lewis J.B. i in. UNIKAJ Badaczy. Aliskiren w skojarzeniu z losartanem w cukrzycy typu 2 i nefropatii // N. Engl. J. Med. - 2008. - Cz. 358 (23). - S. 2433-2446.
16. Jordan J., Engli S., Boye S.W. i in. Bezpośrednie hamowanie reniny za pomocą aliskirenu u otyłych pacjentów z nadciśnieniem tętniczym // Nadciśnienie. - 2007. - Cz. 49. - S. 1-9.
17. Uresin Y., Taylor A., Kilo C. et al. Skuteczność i bezpieczeństwo bezpośredniego inhibitora reniny aliskirenu i ramiprylu samodzielnie lub w skojarzeniu u pacjentów z cukrzycą i nadciśnieniem tętniczym // Renina. Angiotensyna Aldosteron Syst. - 2007. - Cz. 8. - S. 190-198.
18. Oparil S., Yarrows S., Patel S. et al. Skuteczność i bezpieczeństwo łącznego stosowania aliskirenu i walsartanu u pacjentów z nadciśnieniem tętniczym: randomizowana, podwójnie ślepa próba // Lancet. - 2007. - Cz. 370. - S. 221-229.
19. Drummond W., Munger MA, Essop MR. i in. Skuteczność przeciwnadciśnieniowa doustnego bezpośredniego inhibitora reniny aliskirenu jako terapii dodatkowej u pacjentów niereagujących na monoterapię amlodypiną // Clin. nadciśnienie. - 2007. - Cz. 9. - S. 742-750.
20. Chrysant SG, Murray AV, Hoppe UC i in. Długoterminowe bezpieczeństwo, tolerancja i skuteczność aliskirenu w skojarzeniu z walsartanem u pacjentów z nadciśnieniem tętniczym: 6-miesięczna analiza pośrednia // Curr. Med. Rez. Opinia. - 2008. - Cz. 24(4). - S. 1039-1047.
21. Próba aliskirenu w cukrzycy typu 2 z wykorzystaniem punktów końcowych sercowo-nerkowych (WYSOKOŚĆ): uzasadnienie i projekt badania // Przeszczep tarczy nefrolowej. - 2009. - Cz. 24(5). - S. 1663-1671.
22. Stanton AV, Dicker P., O'Brien ET. Monoterapia aliskirenem powoduje największe i najmniejsze obniżenie ciśnienia krwi u pacjentów z odpowiednio wysokim i niskim wyjściowym poziomem PRA // Am. J. Nadciśnienie. - 2009. - Cz. 22. - S. 954-957.
23. Mukhin N. A., Fomin V. V. Aktywność reninowa osocza - czynnik ryzyka i niezależny cel terapii hipotensyjnej: rola aliskirenu // Consilium Medicum. - T. 11. - 2009. - Nr 10. - S. 3-6.
24. Solomon SD, Appelbaum E., Manning W.J. i in. Wpływ bezpośredniego inhibitora reniny aliskirenu, samego lub w połączeniu z losartanem, w porównaniu z losartanem, na masę lewej komory u pacjentów z nadciśnieniem tętniczym i przerostem lewej komory // Próba oceny przerostu lewej komory Aliskirenu (ALLAY). Przedstawiony przez American College of Cardiology. 57. doroczna sesja naukowa, 31 marca 2008 r.
25. Mc. Murray JV, Pitt B., Latini R. et al. Wpływ doustnego bezpośredniego inhibitora reniny aliskirenu u pacjentów z objawową niewydolnością serca // Niewydolność krążenia-serca. - 2008. - Cz. 1. - S.17-24.
26. Nakao N., Yoshimura A., Morita H. et al. Skojarzone leczenie blokera receptora angiotensyny II i inhibitora enzymu konwertującego angiotensynę w niecukrzycowej chorobie nerek (WSPÓŁPRACA): randomizowane badanie kontrolowane // Lancet. - 2003. - Cz. 361 (9352). - S. 117-124.
27. Mann I.F., Schmieder RE, Mc.Queen M. et al. Wyniki dotyczące nerek z telmisartanem, ramiprylem lub obydwoma u osób z wysokim ryzykiem naczyniowym (badanie ONTARGET): wieloośrodkowe, randomizowane, kontrolowane badanie z podwójnie ślepą próbą // Lancet. - 2008. - Cz. 372 (9638). - S. 547-553.
28. Chazova I.E., Fomin V.V., Paltseva E.M. Bezpośredni inhibitor reniny aliskiren – nowe możliwości ochrony nerek w nadciśnieniu tętniczym // Nefrologia Kliniczna. - Nr 1. - 2009. - C. 44-49.
29. Chazova I.E., Fomin V.V. Bezpośredni inhibitor reniny aliskiren: możliwości korekcji zespołu sercowo-nerkowego // Nadciśnienie układowe. - 2009. - Nr 4. - C. 53-58.
30. Diagnostyka i leczenie nadciśnienia tętniczego: zalecenia rosyjskie // Nadciśnienie ogólnoustrojowe. - 2010. - Nr 3. - C. 5-26.
03.07.2012
386 wyświetleń
W przypadku nadciśnienia tętniczego (nadciśnienia tętniczego) zwiększa się ilość enzymu reniny we krwi. Prowadzi to do trwałego i długotrwałego wzrostu ilości białka angiotensyny 2 we krwi i tkankach organizmu.Angiotensyna 2 ma działanie zwężające naczynia krwionośne, sprzyja zatrzymywaniu sodu i wody w organizmie, co prowadzi do wzrostu ciśnienia krwi. Wysoki poziom angiotensyny 2 we krwi i tkankach utrzymujący się przez długi czas powoduje utrzymujący się wzrost ciśnienia krwi, czyli nadciśnienie tętnicze. Inhibitor reniny - lek, który łączy się z reniną, w wyniku czego renina zostaje zneutralizowana i traci aktywność enzymatyczną. To wzajemnie prowadzi do obniżenia poziomu angiotensyny 2 we krwi i tkankach - do obniżenia ciśnienia krwi.
AT2 ma działanie zwężające naczynia krwionośne, sprzyja zatrzymywaniu sodu i wody w organizmie. Prowadzi to do wzrostu i zwiększenia objętości krążącej krwi. Po drugie, następuje wzrost siły skurczów serca. Wszystko to łącznie powoduje wzrost (BP) zarówno skurczowego (górnego), jak i rozkurczowego (dolnego). Im wyższy poziom reniny we krwi, tym wyższy poziom AT2 we krwi, tym wyższe ciśnienie krwi.
Nazywa się sekwencja przemian enzymatycznych: renina + angiotensynogen = angiotensyna 1 + ACE = angiotensyna 2 Układ renina-angiotensyna (RAS) Lub Układ renina-angiotensyna-aldosteron (RAAS). Przez aktywację (zwiększoną aktywność) RAS rozumie się wzrost poziomu reniny, AT2 we krwi.
Wysoki poziom reniny we krwi prowadzi do wzrostu poziomu AT2 we krwi i tkankach. Wysoki poziom AT2 we krwi i tkankach przez długi czas powoduje utrzymujący się wzrost ciśnienia krwi, czyli -.
Spadek poziomu reniny we krwi prowadzi do obniżenia poziomu AT2 we krwi i tkankach - do obniżenia ciśnienia krwi.
Inhibitor reniny- substancja lecznicza, która wchodzi w połączenie z reniną, w wyniku czego renina jest neutralizowana, traci swoją aktywność enzymatyczną, a aktywność enzymatyczna reniny we krwi maleje. Renina związana z inhibitorem reniny traci zdolność do rozszczepiania angiotensynogenu na AT1. Jednocześnie dochodzi do wzajemnie powiązanego obniżenia poziomu AT2 we krwi i tkankach – obniżenie ciśnienia krwi, zmniejszenie aktywności RAS, poprawa ukrwienia, ukrwienia narządów i tkanek ciało.
Aliskiren jest obecnie pierwszym i jedynym inhibitorem reniny, z którym przeprowadzono wszystkie etapy badań klinicznych i który od 2007 roku jest zalecany w leczeniu nadciśnienia tętniczego.
substancja lecznicza Aliskiren produkowane przez przemysł farmaceutyczny pod nazwami handlowymi:
- Rasilez w postaci prostego leku, który zawiera tylko jedną substancję leczniczą - Aliskiren;
- Ko Rasilez w postaci złożonego (złożonego) leku, który zawiera dwa leki: inhibitor reniny Aliskiren i lek moczopędny Hydrochlorotiazyd (saluretyk, diuretyk tiazydowy).
Poniżej możesz umieścić swoją opinię i komentarze na temat stosowania inhibitora reniny Aliskiren w leczeniu nadciśnienia tętniczego.