स्पाइनल वेर्डनिग-हॉफमैन एमियोट्रॉफी एक वंशानुगत बीमारी है। पैथोलॉजी की पृष्ठभूमि के खिलाफ, मोटर न्यूरॉन्स में क्षति नोट की गई है। हॉफमैन की स्पाइनल एमियोट्रॉफी गैर-सेक्स गुणसूत्रों के साथ संचारित होती है। इसके बाद, हम बीमारी, इसकी नैदानिक तस्वीर और इसे खत्म करने के संभावित चिकित्सीय उपायों पर करीब से नज़र डालेंगे।
शब्दावली
स्पाइनल एमियोट्रॉफी कैसे प्रकट होती है, इसके बारे में बात करने से पहले, आइए कुछ अवधारणाओं से परिचित हों। आइए पैथोलॉजी के नाम का विश्लेषण करें। यह दो हिस्सों से मिलकर बना है:
- स्पाइनल - शब्द उल्लंघन के स्थानीयकरण को इंगित करता है। ऐसे में हम रीढ़ की हड्डी में स्थित एक खास तत्व के बारे में बात कर रहे हैं। यह शरीर की सबसे महत्वपूर्ण संरचनाओं में से एक है - रीढ़ की हड्डी।
- एमियोट्रॉफी एक शब्द है जिसमें तीन भाग शामिल हैं: "ए" - विकार, "मायो" - मांसपेशी" और "ट्रॉफी" - पोषण।
इस जानकारी के आधार पर पैथोलॉजी के नाम का अर्थ समझा जा सकता है। स्पाइनल वेर्डनिग-हॉफमैन एमियोट्रॉफी इस प्रकार मांसपेशियों में कुपोषण है। पैथोलॉजी की विशेषता तंतुओं की कमजोरी और मरोड़ की उपस्थिति है।
विरासत
स्पाइनल मस्कुलर एमियोट्रॉफी एक ऑटोसोमल रिसेसिव बीमारी है। यह परिभाषा वंशानुक्रम के प्रकार को इंगित करती है जिसमें किसी गुण का संचरण गैर-लिंग गुणसूत्रों के माध्यम से होता है। इसके अलावा, यह तभी प्रकट होता है जब यह प्रारंभ में माता-पिता दोनों में मौजूद होता है (वे स्वयं बीमार नहीं पड़ सकते हैं)।

रोग का विकास
वयस्कों में स्पाइनल एमियोट्रॉफी नहीं होती है। पैथोलॉजी बच्चों में ही प्रकट होती है। रोग की विशेषता एक घातक पाठ्यक्रम और तेजी से प्रगति है। रीढ़ की हड्डी में बड़ी कोशिकाएं गतिविधियों के समन्वय के लिए जिम्मेदार होती हैं। वे मांसपेशियों की टोन का भी समर्थन करते हैं। जब वे क्षतिग्रस्त हो जाते हैं, तो मांसपेशियों में शिथिलता विकसित हो जाती है।
जन्मजात रूप
स्पाइनल एमियोट्रॉफी के तीन रूप होते हैं। वे पहले लक्षणों के प्रकट होने के समय और प्रक्रिया के विकास की तीव्रता के अनुसार निर्धारित होते हैं। जन्मजात रूप जन्मपूर्व काल में भी शुरू हो सकता है। इस मामले में, गर्भावस्था के बाद के चरणों में भ्रूण की गति कमजोर हो जाती है। उसी समय, प्रसवपूर्व अवधि की शुरुआत में, हलचलें सामान्य सीमा के भीतर थीं। गर्भावस्था का संकल्प ही रोगात्मक हो सकता है। अक्सर, जन्म के बाद पहले कुछ दिनों के भीतर, स्पष्ट मांसपेशी पैरेसिस का पता लगाया जाता है, इसके स्वर में कमी और कण्डरा सजगता के बिगड़ने के साथ। रेट्रोबुलबार (प्रारंभिक) लक्षण भी हो सकते हैं। वे शिशु के कमजोर रोने और सुस्त तरीके से चूसने से प्रकट होते हैं। कुछ मामलों में, पूर्ण एरेफ़्लेक्सिया देखा जाता है। बच्चे की जीभ में कंपन, हाइपोमिया और निगलने की क्षमता में कमी हो सकती है। स्पाइनल एमियोट्रॉफी टैचीकार्डिया के साथ होती है। अक्सर, पैथोलॉजी को कई विकृतियों के साथ जोड़ा जाता है, जो मानस के गठन को धीमा कर देता है। स्पाइनल एमियोट्रॉफी की विशेषता तीव्र गति होती है और 1-1.5 साल में घातक परिणाम के साथ समाप्त हो जाती है।

प्रारंभिक रूप
यह जन्मजात की तुलना में हल्के पाठ्यक्रम की विशेषता है। प्रारंभिक बचपन के रूप को बीमारी की क्लासिक अभिव्यक्ति माना जाता है। इस मामले में स्पाइनल एमियोट्रॉफी डेढ़ साल की उम्र में ही प्रकट होती है।
लगभग सभी मामलों में रोग के लक्षण भोजन विषाक्तता या किसी प्रकार के संक्रामक घाव के बाद पाए जाते हैं। सामान्य रूप से विकसित होने वाला बच्चा पहले से अर्जित मोटर क्षमताओं को जल्दी से खोना शुरू कर देता है। उसका बैठना, खड़ा होना और चलना बंद हो जाता है। सबसे पहले, निचले छोरों में शिथिल पैरेसिस नोट किया जाता है, जो धीरे-धीरे धड़ और भुजाओं तक पहुंचता है। बच्चे की हालत बहुत तेजी से बिगड़ रही है. गर्दन की मांसपेशियों और बल्बर मांसपेशियों में कमजोरी आ जाती है। श्वसन प्रणाली की अपर्याप्तता के परिणामस्वरूप, 4-5 वर्ष की आयु तक निमोनिया प्रकट होता है, फिर मृत्यु हो जाती है। बच्चों में फ्लेसिड पैरेसिस टेंडन सिकुड़न के कारण जटिल होता है। अक्सर स्पाइनल वेर्डनिग-हॉफमैन एमियोट्रॉफी सामान्य हाइपरहाइड्रोसिस के साथ होती है।
पैथोलॉजी की देर से शुरुआत
बीमारी का तीसरा रूप 1.5-2 साल के बाद शुरू होता है। पिछले वाले की तुलना में, यह अपेक्षाकृत आसानी से प्रवाहित होता है। 10 साल तक के बच्चों में चलने-फिरने की क्षमता बनी रहती है। उसके बाद, स्थिति आमतौर पर खराब हो जाती है।

नैदानिक तस्वीर
पैथोलॉजी की विशेषता पैरेसिस है, पहले समीपस्थ निचले छोरों की, और फिर ऊपरी छोरों की। स्पाइनल एमियोट्रॉफी के साथ, वसायुक्त चमड़े के नीचे की परत अच्छी तरह से व्यक्त होती है। बदले में, इससे मांसपेशियों की शिथिलता की पहचान करना मुश्किल हो जाता है। टेंडन रिफ्लेक्सिस काफी पहले ही ख़त्म होने लगते हैं। पैथोलॉजी के लिए, फैली हुई भुजाओं वाली उंगलियों का एक छोटा सा कंपन विशेषता है। हड्डी की विकृति को विशिष्ट माना जाता है, विशेषकर निचले छोरों और उरोस्थि में। बल्बर लक्षण फाइब्रिलर-प्रकार की मरोड़, नरम तालू में पैरेसिस और ग्रसनी प्रतिवर्त में कमी के साथ जीभ की मांसपेशियों के शोष के रूप में प्रकट होते हैं।
फ़ैज़ियो-लोंडे रोग
यह शोष की अभिव्यक्ति का एक विशेष प्रकार है। पैथोलॉजी, एक नियम के रूप में, तीन साल की उम्र तक और कुछ मामलों में किशोरावस्था के दौरान विकसित होने लगती है। इस रोग की विशेषता चबाने वाली मांसपेशियों सहित चेहरे की मांसपेशियों की कमजोरी है। निगलने में कठिनाई और आवाज में बदलाव नोट किया जाता है। पैथोलॉजी जीभ के शोष के साथ होती है, कुछ मामलों में नेत्र रोग प्रकट हो सकता है। रोग बहुत तेजी से बढ़ता है। 6-12 महीने के बाद मृत्यु हो जाती है। चरम सीमाओं में पक्षाघात और पैरेसिस को बल्बर विकारों में जोड़ा जा सकता है। कुछ मामलों में, इन लक्षणों को विकसित होने का समय भी नहीं मिलता है। हालाँकि, एक शव परीक्षा में हमेशा पूर्वकाल रीढ़ की हड्डी के सींगों की कोशिकाओं में एक घाव का पता चलता है।

निदान
जांच के दौरान, पैथोलॉजी को ओपेनहेम के मायोटोनिया से अलग किया जाता है। अधिकांश विशेषज्ञों का मानना है कि यह विकृति विज्ञान कोई स्वतंत्र नोसोलॉजिकल इकाई नहीं है। शोधकर्ताओं के अनुसार, मायोटोनिया ओपेनहेम एक सिंड्रोम है जिसके लिए एक स्पष्ट प्रकार की मांसपेशियों का हाइपोटोनिया प्रमुख अभिव्यक्ति बन जाता है। इस संबंध में, "सुस्त बच्चा" शब्द का हाल ही में व्यापक रूप से उपयोग किया गया है।
अनुसंधान की विधियां: इलेक्ट्रोमायोग्राफी
स्पाइनल एमियोट्रॉफी का पता कई अतिरिक्त अध्ययनों के परिणामों पर आधारित है (प्रारंभिक अभिव्यक्ति और विशिष्ट नैदानिक तस्वीर को छोड़कर)। इनमें से, यह इलेक्ट्रोमोग्राफी पर प्रकाश डालने लायक है। लगभग सभी मामलों में, फासीक्यूलेशन क्षमता की उपस्थिति में आराम के समय बायोइलेक्ट्रिकल सहज गतिविधि का पता लगाया जाता है। स्वैच्छिक संकुचन की पृष्ठभूमि के खिलाफ, "पलिसडे" लय के साथ धीमी प्रकृति की विद्युत गतिविधि का पता लगाया जाता है। यह क्षमता की अवधि और सिंक्रनाइज़ेशन की घटना में वृद्धि का संकेत देता है।

पैथोलॉजिकल अध्ययन
यह आपको पूर्वकाल रीढ़ की हड्डी के सींगों की कोशिकाओं की संख्या में कमी, साथ ही अपक्षयी प्रकार में परिवर्तन की पहचान करने की अनुमति देता है। कपाल नसों के मोटर नाभिक में, ग्रीवा और काठ की मोटाई के क्षेत्र में पैथोलॉजिकल विकार स्पष्ट होते हैं। तंत्रिका अंत के इंट्रामस्क्युलर ज़ोन में, पूर्वकाल की जड़ों में भी परिवर्तन पाए जाते हैं। सामान्य टर्मिनल का गायब होना और अत्यधिक शाखाएं होना है।
जैव रासायनिक विश्लेषण
यह अध्ययन आपको कार्बोहाइड्रेट चयापचय में परिवर्तन की पहचान करने की अनुमति देता है। इस प्रकार, यह पाया गया कि स्पाइनल एमियोट्रॉफी के साथ, रोगियों में ग्लाइकोलाइसिस भ्रूण प्रकार के करीब है। अक्सर, क्रिएटिन-क्रिएटिनिन चयापचय में महत्वपूर्ण परिवर्तन पाए जाते हैं - क्रिएटिन उत्सर्जन में वृद्धि, क्रिएटिनिन रिलीज में कमी। यह भी ध्यान दिया जाना चाहिए कि रक्त सीरम में एंजाइमों की एकाग्रता व्यावहारिक रूप से अपरिवर्तित है।

स्पाइनल एमियोट्रॉफी: उपचार
पैथोलॉजी थेरेपी को व्यायाम चिकित्सा और मालिश की नियुक्ति तक सीमित कर दिया गया है। इन प्रक्रियाओं को नियमित रूप से किया जाना चाहिए। उपचार के कट्टरपंथी तरीके अनुपस्थित हैं। कुछ हद तक, कई दवाएँ लेने से राहत मिल सकती है। विशेष रूप से, विशेषज्ञ "संगविनारिन", "गैलेंटामाइन", "ओक्साज़िल", "प्रोज़ेरिन" जैसे साधनों की सलाह देते हैं। इसके अतिरिक्त, समूह बी के विटामिन निर्धारित हैं। रोग की गंभीर अभिव्यक्तियों के साथ, छोटी खुराक में बार-बार रक्त आधान की सिफारिश की जा सकती है।
वेर्डनिग हॉफमैन की स्पाइनल मस्कुलर एट्रोफी एक दुर्लभ बीमारी है जो मानव न्यूरोमस्कुलर सिस्टम में होती है। यह रोग विभिन्न उत्परिवर्तनों के कारण प्रकट होता है। डॉक्टरों का मानना है कि स्पाइनल एमियोट्रॉफी का मुख्य कारण खराब आनुवंशिकता है।
रोग की गंभीरता के आधार पर रोग के लक्षण नाटकीय रूप से भिन्न होंगे। डॉक्टर सशर्त रूप से स्पाइनल मस्कुलर एट्रोफी को गंभीरता के 4 डिग्री में विभाजित करते हैं। ध्यान दें कि सभी 4 प्रकारों में एक सामान्य विशेषता है - रोगी मानसिक विकास में विफलताओं का अनुभव करता है। किसी भी स्तर पर पेल्विक क्षेत्र में स्थित विभिन्न अंगों की शिथिलता का कारण नहीं बन सकता।
हाथ, पैर और शरीर की मांसपेशियों के शोष पर।
इस बीमारी का मुख्य कारण खराब आनुवंशिकी है। कई वैज्ञानिक अध्ययनों से साबित हुआ है कि पांचवें मानव गुणसूत्र में उत्परिवर्तन के कारण स्पाइनल एट्रोफी प्रकट होती है। जो जीन बदलता है वह मोटर न्यूरॉन्स के सामान्य विकास के लिए जिम्मेदार एक विशेष प्रोटीन के संश्लेषण के लिए जिम्मेदार होता है। इस तथ्य के कारण कि ये न्यूरॉन्स अपने सभी कार्य पूरी तरह से नहीं कर पाते हैं, मनुष्यों में शरीर की मांसपेशियां शोष करने लगती हैं।
डॉक्टरों का कहना है कि बच्चे में यह बीमारी तभी सामने आ सकती है जब पिता और मां दोनों में गलत जीन मौजूद हो। इसके अलावा, डॉक्टरों का कहना है कि पृथ्वी पर हर दूसरा व्यक्ति गलत जीन का वाहक है।
रोग के पहले और दूसरे रूप में लक्षण
वेर्डनिग-हॉफमैन टाइप 1 की स्पाइनल एमियोट्रॉफी अक्सर बच्चे के जीवन के पहले 6 महीनों में दिखाई देती है। कुछ मामलों में, बच्चे के जन्म के दौरान निदान के दौरान बीमारी का पता लगाया जा सकता है। चरण 1 में, बच्चे की मांसपेशियों के तंतुओं की टोन कम हो जाती है, और टेंडन रिफ्लेक्सिस पूरी तरह से अनुपस्थित हो सकते हैं। कभी-कभी निगलने की क्रिया प्रभावित होती है। जीभ, एक नियम के रूप में, भी शोषित होती है। स्टेज 1 में, बच्चे की पाचन क्रिया गड़बड़ा जाती है। इस वजह से भोजन श्वसन पथ में प्रवेश कर सकता है। साँस लेने में भी परेशानी होती है, इस वजह से, फुफ्फुसीय अपर्याप्तता प्रकट हो सकती है। इसके अलावा, पहली डिग्री के स्पाइनल एमियोट्रॉफी वाले बच्चों में, मोटर कौशल का विकास ख़राब होता है और छाती की विकृति देखी जाती है। आँकड़ों के अनुसार, यदि बीमारी जन्म के तुरंत बाद बढ़ने लगती है, तो बच्चा जीवन के पहले 3-4 महीनों में मर जाता है।
दूसरी डिग्री के स्पाइनल एट्रोफी के साथ, बच्चे में जन्म के 6 महीने बाद ही खतरनाक लक्षण विकसित होते हैं। प्रारंभ में, रोगी को जांघ की मांसपेशियों में शोष विकसित हो जाता है। टेंडन रिफ्लेक्स धीरे-धीरे सुस्त हो जाते हैं। चेहरे की मांसपेशियां, एक नियम के रूप में, शोष के अधीन नहीं हैं। कुछ मामलों में, रोगी के हाथों में कंपन होता है और सांस लेने में परेशानी होती है। समय के साथ गर्दन की मांसपेशियां भी कमजोर हो जाती हैं। समय के साथ, रोगी में स्कोलियोसिस या कूल्हे के जोड़ की अव्यवस्था विकसित हो जाती है। ग्रेड 2 स्पाइनल एट्रोफी वाले बच्चे खराब श्वसन क्रिया के कारण रोगजनक संक्रमण के प्रति अधिक संवेदनशील होते हैं। असमय उपचार मिलने पर मरीज की दम घुटने से मौत भी हो सकती है।
: रोग के कारण और उपचार के तरीके।
पीठ के निचले हिस्से को खींचने पर क्या करें? उपचारों के बारे में और जानें.
इसे सही और प्रभावी ढंग से कैसे करें?
तीसरे और चौथे प्रकार के एमियोट्रॉफी में लक्षण
वेर्डनिग-हॉफमैन चरण 3 की स्पाइनल एमियोट्रॉफी, एक नियम के रूप में, किशोरावस्था में प्रकट होती है। इस प्रकार की बीमारी का एक विशिष्ट लक्षण अस्थिर चलना है। यह इस तथ्य से समझाया गया है कि पैरों की मांसपेशियों की टोन तेजी से कम हो जाती है। यह इस तथ्य के कारण है कि मांसपेशी फाइबर पतले हो जाते हैं। रोगी अक्सर लड़खड़ा सकता है या गिर भी सकता है। समय के साथ, रोगी पूरी तरह से चलना बंद कर सकता है। कभी-कभी हाथों की मांसपेशियों में शोष दिखाई देता है। रोगी के चेहरे के भाव भी बिगड़ जाते हैं। कंकाल में परिवर्तन हो रहा है। रोगी की छाती विकृत हो जाती है तथा जोड़ों का कार्य बाधित हो जाता है। दुर्लभ मामलों में, रोगी को सामान्य साँस लेने में परेशानी होती है।
वेर्डनिग-हॉफमैन चरण 4 की स्पाइनल एमियोट्रॉफी, एक नियम के रूप में, वयस्कता में प्रकट होती है। रोग के इस चरण में एक विशिष्ट लक्षण पैरों में कमजोरी का आना है। रोगी को मांसपेशियों में गंभीर दर्द की शिकायत हो सकती है। समय के साथ, पैरों की मांसपेशियां कमजोर होने लगती हैं और प्रतिक्रियाएं सुस्त हो जाती हैं। स्पाइनल मस्कुलर एट्रोफी की प्रगति इस तथ्य की ओर ले जाती है कि रोगी चलना बंद कर देता है। ग्रेड 4 पर, श्वसन क्रिया नहीं बदलती है। हाथों की मांसपेशियां पूरी तरह क्रियाशील होती हैं। डॉक्टरों का मानना है कि बीमारी का प्रकार 4 सौम्य है।
पैथोलॉजी का निदान और उपचार
यदि किसी व्यक्ति में स्पाइनल एट्रोफी के पहले लक्षण दिखाई देते हैं, तो उसे एक व्यापक निदान निर्धारित किया जाता है। सबसे पहले, रोगी को इलेक्ट्रोन्यूरोमायोग्राफी से गुजरना होगा। इस प्रक्रिया से, डॉक्टर न्यूरॉन्स के कामकाज में असामान्यताओं का पता लगा सकते हैं। उसके बाद, रोगी को आनुवंशिक अध्ययन से गुजरना होगा। इस प्रक्रिया के दौरान डॉक्टर मरीज के डीएनए से प्राप्त डेटा का अध्ययन करते हैं। आनुवंशिक अनुसंधान से गुणसूत्र 5 में उत्परिवर्तन का पता चलता है।
यदि किसी महिला के करीबी रिश्तेदारों को यह बीमारी है, तो गर्भावस्था के दौरान उसे विशेष प्रसवपूर्व निदान से गुजरना चाहिए। यदि भ्रूण में कोई विकृति पाई जाती है, तो गर्भावस्था को जबरन समाप्त करना आवश्यक हो सकता है।
स्पाइनल एट्रोफी का इलाज नहीं किया जाता है। हालांकि, विशेष दवाओं की मदद से रोगी की स्थिति को कम करना संभव है जो तंत्रिका ऊतकों में चयापचय प्रक्रियाओं में सुधार करते हैं। थेरेपी को बी विटामिन, एनाबॉलिक स्टेरॉयड का एक कोर्स, व्यायाम चिकित्सा और फिजियोथेरेपी के साथ पूरक किया जाता है।
अतिरिक्त स्रोत:
- 1. सु वाई.एन., हंग सी.सी., लिन एस.वाई., चेन एफ.वाई., चेर्न जे.पी., त्साई सी. स्पाइनल मस्कुलर एट्रोफी (एसएमए) के लिए स्क्रीनिंग // पब्लिक लाइब्रेरी ऑफ साइंस
- 2. सुगरमैन ई. ए., नागन एन., झू एच., अकमेव वी. आर., झोउ जेड., रोहल्फ़्स ई. एम., फ्लिन के., हेंड्रिकसन बी. सी., शोल टी., सिरको-ओसाडसा डी. ए., एलीटो बी. ए. स्पाइनल मस्कुलर एट्रोफी के लिए पैन-एथनिक कैरियर स्क्रीनिंग और प्रसवपूर्व निदान: नैदानिक प्रयोगशाला विश्लेषण यूरोपीय जर्नल ऑफ ह्यूमन जेनेटिक्स।
- 3. प्रकार I, II, III, IV की स्पाइनल एमियोट्रॉफी… // मेडिकल जेनेटिक रिसर्च सेंटर के आणविक आनुवंशिकी केंद्र।


सभी मौजूदा और विज्ञान के लिए ज्ञात स्पाइनल मस्कुलर एट्रोफी में से वेर्डनिग-हॉफमैन एमियोट्रॉफीसबसे भारी किस्म है.
इस बीमारी की व्यापकता अब प्रति 7-11 हजार नवजात शिशुओं पर लगभग 1 मामला है।
इस बीमारी का कारण बनने वाला जीन हर 50वें व्यक्ति में मौजूद होता है।
हालाँकि, ऑटोसोमल रिसेसिव इनहेरिटेंस के कारण, बच्चे में विकार तभी होता है जब माता-पिता दोनों के पास यह आनुवंशिक जानकारी होती है।
इसलिए, इस मामले में, संभावना है कि बच्चा किसी विकृति के साथ पैदा होगा, लगभग 25% है।
क्या ऐसी बीमारी से निपटना संभव है, या कम से कम लक्षणों की प्रगति को रोकना संभव है, हम इस लेख में बताएंगे।
वेर्डनिग-हॉफमैन एमियोट्रॉफी क्या है?
स्पाइनल एमियोट्रॉफी टाइप 1 या, दूसरे शब्दों में, वेर्डनिग-हॉफमैन स्पाइनल एमियोट्रॉफी- यह तंत्रिका तंत्र की एक विशेष बीमारी है, जो विरासत में मिली है (अक्सर माता-पिता दोनों से)। यह विकृति शरीर की लगभग संपूर्ण मांसपेशी प्रणाली में मांसपेशियों की कमजोरी की उपस्थिति की विशेषता है। ऐसी बीमारी से पीड़ित बच्चा न तो खुद बैठ सकता है, न हिल सकता है और न ही अपनी सेवा कर सकता है।
दुर्भाग्य से, दुनिया में इस प्रकार की बीमारी का कोई इलाज नहीं है। हमारे समय में डॉक्टर जो अधिकतम पेशकश कर सकते हैं वह है प्रसव पूर्व निदान। इस तरह की जांच से परिवार में बीमार बच्चे के जन्म से बचने में मदद मिलती है।
पैथोलॉजी को इसका नाम दो वैज्ञानिकों के नाम पर मिला जिन्होंने पहली बार 19वीं सदी के अंत में इसका वर्णन किया था। वर्तमान में, स्पाइनल एमियोट्रॉफी की अवधारणा रोग के कई रूपों को संदर्भित करती है जो चिकित्सकीय रूप से भिन्न होते हैं। लेकिन वे सभी उसी आनुवंशिक दोष से जुड़े हैं जो बच्चे के माता-पिता में है।
रोग की नैदानिक तस्वीर
स्पाइनल एमियोट्रॉफी के कई रूप और किस्में हैं, जिनमें से प्रत्येक विशिष्ट लक्षणों की शुरुआत की उम्र, रोग के पाठ्यक्रम की गंभीरता और रोगियों की जीवन प्रत्याशा में भिन्न होता है।
आम तौर पर यह विकृति विकलांगता की ओर ले जाती है, क्योंकि शरीर की मोटर प्रणाली परेशान है, और रोगी स्वतंत्र रूप से चलने या स्वतंत्र रूप से अपनी सेवा करने में सक्षम नहीं है। गंभीर नैदानिक स्थितियों में, रोजमर्रा की जिंदगी में निरंतर चिकित्सा पर्यवेक्षण आवश्यक हो सकता है।

व्हीलचेयर, वॉकर, बैसाखी, बेंत ऐसे रोगी को चलने-फिरने में मदद करते हैं। ऐसी बीमारी घातक परिणाम तभी दे सकती है जब श्वसन और हृदय प्रणाली (निमोनिया और हृदय विफलता के साथ) से जटिलताएं सामने आती हैं।
संवेदनशील तंत्रिका तंतु विकृति विज्ञान के प्रभाव में नहीं आते, इसलिए बच्चे में सभी प्रकार की संवेदनशीलता बनी रहती है। बुद्धि और मानसिक कार्य भी प्रभावित नहीं होते हैं, इसलिए सीखते समय, बच्चा सामान्य रूप से जानकारी को समझता और आत्मसात करता है।
रोग वर्गीकरण
जिस उम्र में रोग के लक्षण दिखाई देते हैं, उसके आधार पर वेर्डनिग-हॉफमैन एमियोट्रॉफी को कई प्रकारों में विभाजित किया जाता है:
- पैथोलॉजी का जन्मजात रूप. परिवर्तनों के प्रकट होने की अनुमानित आयु: 0 से 6 महीने तक। आमतौर पर कमजोर अंतर्गर्भाशयी भ्रूण गतिविधि की विशेषता होती है। जन्मजात रूप के साथ, शिशु के जीवन के पहले दिनों से ही मांसपेशी हाइपोटेंशन देखा जाता है। थोड़े समय के भीतर, गहरी सजगता फीकी पड़ जाती है: बच्चा कमजोर रूप से रोता है, मां का दूध या निप्पल खराब तरीके से चूसता है, अपना सिर नहीं पकड़ पाता है। कभी-कभी ऐसा होता है कि ये लक्षण थोड़ी देर से दिखाई देते हैं, इसलिए बच्चा अपना सिर पकड़ना और बैठना सीख सकता है, लेकिन उल्लंघन होने के कारण उसमें ये कौशल विकसित नहीं हो पाएंगे। इसके अलावा, जन्मजात रूप के साथ बल्ब संबंधी विकार, ग्रसनी प्रतिवर्त में कमी और जीभ की फेशियल मरोड़ भी हो सकती है। जन्मजात रूप को सबसे घातक माना जाता है और इसमें अक्सर ओलिगोफ्रेनिया, छाती की विकृति और स्कोलियोसिस की 4 डिग्री भी शामिल हो सकती है। श्वसन प्रणाली की तीव्र गतिहीनता और पक्षाघात से श्वसन विफलता और बाद में मृत्यु हो जाती है;
- प्रारंभिक बचपन का स्वरूप.इस प्रकार की विकृति के साथ, पहले लक्षण 6 महीने के बाद दिखाई दे सकते हैं। इस समय तक बच्चों का शारीरिक और मानसिक विकास सामान्य हो जाता है। वे धीरे-धीरे पहला प्राकृतिक कौशल हासिल करना शुरू करते हैं, जैसे सिर पकड़ने, खड़े होने, बैठने और पलटने की क्षमता। ज्यादातर मामलों में, इस प्रकार की बीमारी से बच्चे कभी चलना नहीं सीख पाएंगे। प्रारंभिक चरण में, पैरेसिस निचले छोरों में होता है, फिर वे ऊपरी छोरों और संपूर्ण मांसपेशियों में तेजी से विकसित होते हैं। मांसपेशियों में हाइपोटेंशन शुरू हो जाता है, गहरी प्रतिक्रियाएँ फीकी पड़ जाती हैं, उंगलियाँ कांपती हैं, अनैच्छिक मांसपेशी संकुचन दिखाई दे सकते हैं। बाद के चरणों में, बल्बर विकार और श्वसन विफलता (प्रगतिशील) सभी लक्षणों में जुड़ जाते हैं। रोग का यह रूप जन्मजात प्रकार की तुलना में अधिक धीरे-धीरे बढ़ता है। मरीज़ 15 साल तक जीवित रह सकते हैं;
- कुगेलबर्ग-वेलैंडर एमियोट्रॉफी।स्पाइनल एमियोट्रॉफी के सभी रूपों में सबसे सौम्य। लक्षण 2 साल के बाद दिखाई देते हैं, कभी-कभी 15वें से 30वें साल के बीच। इस रूप के साथ, विकास में कोई मानसिक मंदता नहीं होती है, काफी लंबे समय तक रोगी स्वतंत्र रूप से चलने में सक्षम होते हैं। अनेक लोग पूर्ण स्व-सेवा पर परिपक्व बुढ़ापे तक जीवित रहते हैं।
रोग के जोखिम कारक और कारण
चूँकि वेर्डनिग-हॉफमैन स्पाइनल एमियोट्रॉफी एक वंशानुगत बीमारी है इसकी घटना के कारण रोगी के माता-पिता दोनों के आनुवंशिक कोड में निहित हैं. समस्या पांचवें गुणसूत्र में है, जो आनुवंशिक उत्परिवर्तन से गुजरता है।
उस जीन को उत्परिवर्तित करता है जो एसएमएन प्रोटीन के उत्पादन के लिए जिम्मेदार है. एक स्वस्थ शरीर में, इस प्रोटीन का संश्लेषण मोटर न्यूरॉन्स के सामान्य विकास को सुनिश्चित करता है। यदि इसमें उत्परिवर्तन हुआ है, तो मोटर न्यूरॉन्स ढहने लगते हैं, जिससे तंत्रिका फाइबर से मांसपेशियों तक आवेगों के संचरण में व्यवधान होता है। नतीजतन, मांसपेशियां काम नहीं करतीं। इसीलिए मोटर शोष और सामान्य रूप से चलने में असमर्थता होती है।

विकारों के साथ जीन एक ऑटोसोमल रिसेसिव इनहेरिटेंस पैटर्न है. इसका मतलब यह है कि बीमारी के विकसित होने के लिए, माता-पिता दोनों के दो उत्परिवर्तित जीनों का मेल आवश्यक है। वे। वास्तव में, पिता और माता दोनों को पैथोलॉजी वाले जीन के वाहक होने चाहिए।
साथ ही, वे बीमार नहीं हैं, क्योंकि उनके पास एक प्रमुख स्वस्थ जीन है (यह जीन की जोड़ी के कारण भी है)। यदि शिशु के पिता और माता दोनों में विकृति वाला जीन है, तो बच्चे के विकारों के साथ पैदा होने का जोखिम 25% है।
वीडियो: "स्पाइनल मस्कुलर एट्रोफी क्या है?"
रोग के निदान के तरीके
जब इस प्रकार की बीमारी का निदान करने की बात आती है, तो यह ध्यान में रखा जाना चाहिए कि परीक्षा आयोजित करने वाले न्यूरोलॉजिस्ट के लिए, वह उम्र बहुत महत्वपूर्ण है जिस पर बच्चे में पहले लक्षण दिखाई देते हैं।
भी लक्षणों के विकास की गतिशीलता महत्वपूर्ण है, न्यूरोलॉजिकल स्थिति डेटा (यानी, सामान्य संवेदनशीलता की पृष्ठभूमि के खिलाफ परिधीय प्रकार के मोटर विकारों की उपस्थिति / अनुपस्थिति), अतिरिक्त जन्मजात विसंगतियों और हड्डी विकृति (स्क्लिओसिस, किफोसिस, लॉर्डोसिस, टॉरिसोलिस) की उपस्थिति / अनुपस्थिति।
रोग के जन्मजात प्रकार का पता एक नवजातविज्ञानी द्वारा लगाया जा सकता है. परीक्षा मायोपैथी, मस्कुलर डिस्ट्रॉफी (प्रगतिशील), एमियोट्रोफिक लेटरल स्क्लेरोसिस, पोलियोमाइलाइटिस, सेरेब्रल पाल्सी आदि के लिए की जाती है। यदि निदान के लिए सबसे सटीक पुष्टि की आवश्यकता होती है, तो इलेक्ट्रोन्यूरोमायोग्राफी (न्यूरोमस्कुलर उपकरण का अध्ययन) का भी उपयोग किया जाता है।
 अंतिम निदान मांसपेशियों के जीव विज्ञान पर डेटा प्राप्त करने और आनुवंशिक स्थिति का अध्ययन करने के बाद ही स्थापित किया जाता है।. डीएनए विश्लेषण का अध्ययन आनुवंशिकीविदों को जीन विपथन की विषमयुग्मजी गाड़ी का पता लगाने की अनुमति देता है (अगली गर्भावस्था की योजना बनाते समय महत्वपूर्ण)। एसएमए लोकस के जीनों की संख्या का एक मात्रात्मक विश्लेषण भी किया जाता है (आपको माता-पिता में एक पैथोलॉजिकल जीन की उपस्थिति की गणना करने की अनुमति देता है।
अंतिम निदान मांसपेशियों के जीव विज्ञान पर डेटा प्राप्त करने और आनुवंशिक स्थिति का अध्ययन करने के बाद ही स्थापित किया जाता है।. डीएनए विश्लेषण का अध्ययन आनुवंशिकीविदों को जीन विपथन की विषमयुग्मजी गाड़ी का पता लगाने की अनुमति देता है (अगली गर्भावस्था की योजना बनाते समय महत्वपूर्ण)। एसएमए लोकस के जीनों की संख्या का एक मात्रात्मक विश्लेषण भी किया जाता है (आपको माता-पिता में एक पैथोलॉजिकल जीन की उपस्थिति की गणना करने की अनुमति देता है।
प्रसव पूर्व डीएनए परीक्षण से वेर्डिंग-हॉफमैन रोग वाले बच्चे के जन्म की संभावना को कम करने में मदद मिल सकती है। लेकिन यहां कठिनाई इस तथ्य में निहित है कि डीएनए सामग्री प्राप्त करने के लिए प्रसव पूर्व निदान (कोरियोनिक बायोप्सी, कॉर्डोसेन्टेसिस, एमनियोसेंटेसिस) के आक्रामक तरीकों का उपयोग किया जाता है।
यदि गर्भाशय में रोग की पुष्टि हो जाती है, तो यह वर्तमान गर्भावस्था के कृत्रिम समापन का संकेत होगा।
रोग का उपचार
आधुनिक चिकित्सा, दुर्भाग्य से, अभी तक ऐसी दवाएं विकसित नहीं कर पाई है जो विभिन्न प्रकार के आनुवंशिक उत्परिवर्तनों का सामना कर सकें, इसलिए, कोई ऐसा उपचार पाठ्यक्रम मौजूद नहीं है जो वेर्डनिग-हॉफमैन एमियोट्रॉफी को हरा सके। ऐसी कई गतिविधियां हैं जो प्रगतिशील मांसपेशी शोष को धीमा कर सकती हैं, लेकिन चमत्कार की उम्मीद नहीं की जा सकती है।
दवाएं
दवाइयाँ केवल रोगी की स्थिति को कमजोर कर सकता है, कुछ हद तक उसके तंत्रिका तंत्र का समर्थन और पोषण करते हैं, लेकिन वे रोग के विकास के कारण को नहीं हरा सकते हैं।
स्पाइनल एमियोट्रॉफी के साथ, निम्नलिखित दवाओं का उपयोग किया जाता है:
- सेरेब्रोलिसिन, साइटोफ्लेविन, ग्लूटामिक एसिड, एटीपी, कार्निटाइन क्लोराइड, मेथिओनिन, पोटेशियम ऑरोटेटऔर अन्य - ये दवाएं कुछ हद तक मांसपेशियों और तंत्रिका ऊतकों के चयापचय में सुधार करती हैं, इसलिए उन्हें आवधिक पाठ्यक्रम सेवन के लिए निर्धारित किया जाता है;
- बी विटामिन(न्यूरोविटन, मिल्गामा);
- उपचय स्टेरॉइड(रेटाबोलिल, नेरोबोल);
- प्रोज़ेरिन, न्यूरोमिडिन, डिबाज़ोल- ये दवाएं मांसपेशियों की चालकता में कुछ हद तक सुधार कर सकती हैं।

व्यायाम, मालिश, व्यायाम चिकित्सा
और क्या आप जानते हैं कि...
अगला तथ्य
रोगी की स्थिति को कम करने के लिए शारीरिक व्यायाम और मालिश का भी उपयोग किया जाता है।. हालाँकि, ऐसी गंभीर विकृति के मामले में, व्यायाम चिकित्सा और मालिश आपके डॉक्टर द्वारा निर्धारित की जानी चाहिए और किसी विशेषज्ञ की सख्त निगरानी में की जानी चाहिए (यही बात फिजियोथेरेपी पर भी लागू होती है)। यहां किसी भी बुनियादी व्यायाम चिकित्सा परिसरों का उपयोग नहीं किया जा सकता है, केवल पैथोलॉजी के विकास के स्तर, लक्षण लक्षणों और रोग के चरण के अनुसार डॉक्टर द्वारा व्यक्तिगत रूप से चुना जाता है।
वीडियो: "जीन थेरेपी से स्पाइनल मस्कुलर एट्रोफी का इलाज"
घर पर इलाज
इस मामले में डॉक्टर के परामर्श और पर्यवेक्षण के बिना घर पर अनधिकृत उपचार सख्त वर्जित है।. घर पर निवारक उपाय केवल तभी लागू किए जा सकते हैं जब डॉक्टर ने अनुमति दी हो। एक विशेषज्ञ आपको कुछ सरल व्यायाम बता सकता है जिन्हें आप घर पर कर सकते हैं। यदि ऐसी कोई अनुशंसाएँ नहीं होतीं, तो स्वयं कुछ करने की सख्त मनाही होती।
रोग प्रतिरक्षण
इस तथ्य के आधार पर कि वेर्डिंग-हॉफमैन एमियोट्रॉफी एक वंशानुगत बीमारी है इसके लिए कोई निवारक उपाय नहीं हैं. दुर्भाग्य से, यदि प्रसवपूर्व अवधि में भी शिशु की विकृति का पता चला था, तो असफल गर्भावस्था के कृत्रिम समापन का सहारा लेना उचित होगा। इस तरह की बीमारी का अभी तक न तो इलाज खोजा जा सका है और न ही रोकथाम।
रोग का पूर्वानुमान
दुर्भाग्य से, इस विकृति का पूर्वानुमान बेहद प्रतिकूल है।. ठीक होने की कोई संभावना नहीं है और न ही हो सकती है। यदि कोई बच्चा एमियोट्रॉफी के जन्मजात रूप के साथ पैदा हुआ है, तो उसकी मृत्यु 6 महीने से 2 साल की उम्र के बीच हो जाएगी। बाद के प्रकार की बीमारी से रोगी का जीवन लम्बा हो जाता है, लेकिन मृत्यु की संभावना बहुत अधिक रहती है।
निष्कर्ष
आधुनिक चिकित्सा कई ऐसी बीमारियों को जानती है जिनका कोई भी तरीका और कोई भी दवा सामना नहीं कर सकती। स्पाइनल वेर्डनिग-हॉफमैन एमियोट्रॉफीउन्हीं में से एक है। इस विकृति की आनुवंशिक उत्पत्ति ने हमें ज्ञात तरीकों से इसका इलाज करना असंभव बना दिया।
कुछ और महत्वपूर्ण बिंदु भी हैं जिन पर आपको विचार करना चाहिए यदि आपको इस बीमारी के साथ बच्चा होने का खतरा है:
- स्पाइनल वेर्डनिग-हॉफमैन एमियोट्रॉफी की कई किस्में हैं: जन्मजात, प्रारंभिक और कुगेलबर्ग-वेलैंडर एमियोट्रॉफी। जन्मजात रूप से प्रारंभिक मृत्यु हो जाती है (लगभग 6 महीने से 2 वर्ष की आयु में), प्रारंभिक और देर से होने वाला रूप रोगी के जीवन को लम्बा खींच सकता है, लेकिन किसी भी तरह से ठीक होने की कोई संभावना नहीं होती है;
- पैथोलॉजी विकसित होने का जोखिम तभी प्रकट हो सकता है जब माता-पिता दोनों में उत्परिवर्तित जीन हो. केवल इस शर्त पर कि माता और पिता दोनों (जो बीमार नहीं दिखते, बल्कि केवल जीन के वाहक हैं) आनुवंशिक श्रृंखला में विकारों के स्वामी हैं, क्या कोई बच्चा वेर्डनिग-हॉफमैन रोग (25% संभावना) के साथ पैदा हो सकता है;
- आनुवंशिक बीमारी का इलाज करना असंभव है, ठीक उसी तरह जैसे रोकथाम करना असंभव है।. दवाएँ केवल रोगी के लक्षणों को दूर करने और शरीर में टोन बनाए रखने के लिए निर्धारित की जाती हैं। व्यायाम चिकित्सा और मालिश केवल उपस्थित चिकित्सक द्वारा निर्धारित की जाती है, केवल उसके द्वारा और किसी और द्वारा नहीं, क्योंकि रोग गंभीर है और सख्त पर्यवेक्षण की आवश्यकता है। यदि प्रसवपूर्व निदान से पहले ही यह तथ्य सामने आ गया है कि बच्चा किसी विकृति के साथ पैदा होगा, तो डॉक्टर गर्भावस्था के कृत्रिम समापन का सहारा लेने की सलाह देते हैं।
परीक्षा पास करें!
वेर्डनिग-हॉफमैन का स्पाइनल मस्कुलर एट्रोफी एक प्रगतिशील न्यूरोमस्कुलर रोगविज्ञान है जो विरासत में मिला है। यह रोग धड़, निचले छोरों और गर्दन की समीपस्थ धारीदार मांसपेशियों को नुकसान पहुंचाता है।
कारण
यह एक वंशानुगत विकृति है, जो पर आधारित है मानव गुणसूत्र पर उत्परिवर्तन 5. परिणामस्वरूप, एसएमएन प्रोटीन का संश्लेषण, जो मोटर न्यूरॉन्स के सामान्य विकास के लिए आवश्यक है, बाधित हो जाता है।
यह रोग दोषपूर्ण तंत्रिका कोशिकाओं के विनाश या विकास की ओर ले जाता है जो मांसपेशियों के तंतुओं तक आवेगों को संचारित करने में असमर्थ होते हैं। इसलिए, आंतरिक मांसपेशियां काम करना बंद कर देती हैं, शोष विकसित होता है।
आंकड़ों के मुताबिक, हर दूसरा व्यक्ति एक पैथोलॉजिकल जीन का वाहक होता है।
उत्परिवर्तित जीन है ऑटोसोमल रिसेसिव इनहेरिटेंस पैटर्न- रोग के विकास के लिए माता और पिता के दो गलत गुणसूत्रों का मेल आवश्यक है।

एक बीमार बच्चा केवल उन माता-पिता से पैदा होता है जिनमें पैथोलॉजिकल जीन होता है। हालाँकि, माता और पिता में एक स्वस्थ प्रमुख जीन होता है, इसलिए उनमें विकृति के लक्षण नहीं दिखते हैं।
रोग की नैदानिक तस्वीर
एसएमए के पाठ्यक्रम के लक्षण और गंभीरता रोग के पहले लक्षणों के प्रकट होने के समय से निर्धारित होती है। इसलिए, डॉक्टर 3 प्रकार की बीमारियों में अंतर करते हैं:
- जन्मजात रूप;
- बचपन;
- देर से फार्म.

प्रत्येक रूप पर अधिक विस्तार से विचार करना उचित है।
जन्मजात रूप की विशेषताएं
फ्लेसीसिड पैरेसिस वाले बच्चों का जन्म विशेषता है। नवजात शिशुओं में सामान्यीकृत मांसपेशी हाइपोटोनिया, गहरी सजगता की अनुपस्थिति का निदान किया जाता है। बल्ब संबंधी विकार इस तथ्य को जन्म देते हैं कि बच्चा धीरे से स्तन चूसता है, चुपचाप चिल्लाता है, उसकी ग्रसनी प्रतिवर्त कम हो जाती है। समय के साथ, बच्चे को डायाफ्रामिक मांसपेशी के पैरेसिस का निदान किया जाता है।
वेर्डनिग-हॉफमैन स्पाइनल एमियोट्रॉफी के लक्षण 10,000 नवजात शिशुओं में से 1 में पाए जाते हैं।
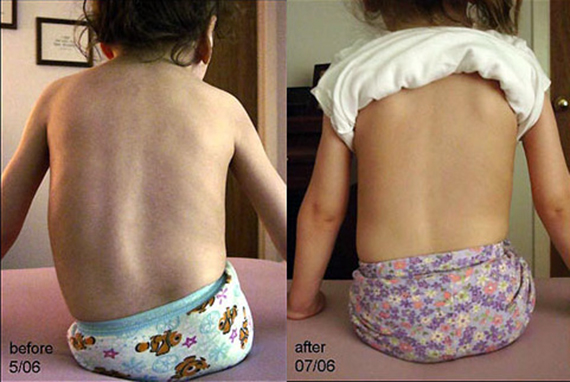
यह रोग अक्सर जुड़ा रहता है हड्डी और जोड़ की विकृति(फ़नल स्टर्नम, स्कोलियोसिस, संयुक्त सिकुड़न)। स्थैतिक और लोकोमोटर कार्यों का धीमा विकास विशेषता है, इसलिए केवल सीमित संख्या में बच्चे ही अपना सिर अपने आप पकड़ने में सक्षम होते हैं। हालाँकि, यह कौशल जल्दी ही वापस आ सकता है।

स्पाइनल मायोपैथी के जन्मजात रूप वाले कई रोगियों में बुद्धि, विकृतियां (क्लबफुट, हाइड्रोसिफ़लस, हेमांगीओमा, क्रिप्टोर्चिडिज़्म) कम हो गई हैं।
रोग तेजी से विकसित होता है और एक घातक पाठ्यक्रम की विशेषता है। मरीज़ शायद ही कभी 9 वर्ष से अधिक जीवित रहते हैं। मृत्यु का कारण दैहिक विकृति है।
हॉफमैन-वेर्डनिग एमियोट्रॉफी के जन्मजात रूप से पीड़ित 50% से अधिक बच्चे 2 साल तक जीवित नहीं रह पाते हैं।
प्रारंभिक बचपन के लक्षण
सिंड्रोम की विशेषता 6 महीने से अधिक उम्र के शिशुओं में विकृति विज्ञान के पहले लक्षणों का विकास है। उसी समय, रोगियों का मोटर विकास संतोषजनक होता है: वे अपना सिर पकड़ने, बैठने और कभी-कभी खड़े होने में सक्षम होते हैं। नशा या किसी संक्रामक रोग की पृष्ठभूमि के विरुद्ध रोग का सूक्ष्म विकास विशेषता है।

पेशीविकृतिइससे पैरों में ढीलापन आ जाता है, जो तेजी से धड़ और भुजाओं की मांसपेशियों तक फैल जाता है। यह मांसपेशियों की टोन, गहरी सजगता में कमी को भड़काता है। बाद के चरणों में, बच्चे को सामान्यीकृत मांसपेशी हाइपोटेंशन, बल्बर पक्षाघात का निदान किया जाता है।
पैथोलॉजी के लिए पूर्वानुमान प्रतिकूल है - बीमारी का एक घातक कोर्स है, बच्चे शायद ही कभी 15 साल तक जीवित रहते हैं।
देर से फार्म
पैथोलॉजी के पहले लक्षण लोकोमोटर और स्थैतिक कार्यों के पूर्ण गठन के बाद दिखाई देते हैं। इसलिए, कई बच्चे अपने आप दौड़ने और चलने में सक्षम होते हैं। विशेषता मायोपैथी का क्रमिक विकास है, जो बच्चे की अजीब और अनिश्चित गतिविधियों में प्रकट होती है। धीरे-धीरे, चाल बदलने लगती है - बच्चे घड़ी की गुड़िया की तरह चलते हैं, लगातार अपने घुटनों को झुकाते हैं।

फ्लेसीड पैरेसिस शुरू में निचले छोरों में स्थानीयकृत होता है, लेकिन धीरे-धीरे धड़ और भुजाओं की मांसपेशियों तक फैल जाता है। बच्चे में अच्छी तरह से विकसित चमड़े के नीचे की वसा होती है, इसलिए मांसपेशी फाइबर का शोष शायद ही ध्यान देने योग्य होता है। धीरे-धीरे, रोगी में बल्बर लक्षण विकसित होते हैं, उंगलियों में कंपन होता है।
एसएमए के शुरुआती चरणों में, गहरी प्रतिक्रियाएँ फीकी पड़ जाती हैं और छाती विकृत होने लगती है।
रोग का घातक रूप है, लेकिन लक्षण पिछले रूपों की तुलना में अधिक धीरे-धीरे विकसित होते हैं। बच्चे केवल 10 वर्ष की आयु तक स्वतंत्र रूप से चलने की क्षमता खो देते हैं। मृत्यु आमतौर पर 30 वर्ष की आयु से पहले होती है।
| एसएमए प्रकार | रोग घोषणापत्र | अधिकतम कार्य | मृत्यु की आयु |
| जन्मजात रूप | पहले लक्षण 6 महीने से कम उम्र के बच्चों में विकसित होते हैं। | बच्चा हिलने-डुलने, सिर पकड़ने, बैठने में असमर्थ है | कई मरीज़ 2 साल की उम्र से पहले मर जाते हैं लेकिन 9 साल तक जीवित रह सकते हैं |
| प्रारंभिक बचपन का स्वरूप | लक्षण 7 से 12 महीने के बीच शुरू होते हैं | रोगी बैठ सकता है, खड़ा हो सकता है, लेकिन धीरे-धीरे उसकी कार्यक्षमता कम हो जाती है | 14-15 साल का |
| देर से फार्म | 1 वर्ष से अधिक उम्र के बच्चों में रोग के लक्षण विकसित होते हैं | बच्चा खड़ा होता है और चलता है | उम्र 20 से 30 साल तक |
निदान उपाय
निदान के लिए पैथोलॉजी के पहले लक्षणों की उम्र, विकास की गतिशीलता और रोगी की न्यूरोलॉजिकल स्थिति (संवेदनशीलता के संरक्षण की पृष्ठभूमि के खिलाफ मोटर कार्यों में गड़बड़ी), हड्डी की विकृति और जन्मजात विसंगतियों की उपस्थिति का बहुत महत्व है। एसएमए के जन्मजात रूप का निदान आमतौर पर नियोनेटोलॉजिस्ट द्वारा किया जाता है।
व्यापक निदान में निम्नलिखित गतिविधियाँ शामिल हैं:

एसएमए वाले बच्चे के जोखिम को कम करने के लिए प्रसव पूर्व डीएनए परीक्षण की सिफारिश की जाती है। हालाँकि, निदान के लिए सामग्री केवल आक्रामक तकनीकों (एमनियोसेंटेसिस, कोरियोनिक बायोप्सी, कॉर्डोसेन्टेसिस) के माध्यम से प्राप्त करना संभव है। यदि गर्भाशय में एमियोट्रॉफी का निदान किया गया था, तो गर्भावस्था की समाप्ति का संकेत दिया गया है।

रोग के उपचार की विशेषताएं
स्पाइनल मायोपैथी एक लाइलाज विकृति है, इसलिए उपचार केवल रोगी की स्थिति को कम कर सकता है। इस प्रयोजन के लिए, निम्नलिखित दवाओं का उपयोग किया जाता है:
- उपचय स्टेरॉइड;
- बी विटामिन;
- इसका मतलब है कि मांसपेशियों और न्यूरॉन्स के चयापचय में सुधार;
- दवाएं जो न्यूरोमस्कुलर चालन में सुधार करती हैं।
इसके अतिरिक्त, एक पाठ्यक्रम निर्धारित है मालिश और व्यायाम चिकित्सा, फिजियोथेरेपी (विद्युत मांसपेशी उत्तेजना, ऑक्सीजन थेरेपी), आर्थोपेडिक सुधार। रोगी को आहार-विहार का पालन करना चाहिए।

यदि श्वसन विफलता विकसित होती है, तो सांस लेने को बहाल करने के लिए बीमार बच्चे को वेंटिलेटर से जोड़ा जाता है। निगलने की प्रतिक्रिया के स्पष्ट उल्लंघन के साथ, गैस्ट्रोस्टोमी फीडिंग ट्यूब के उपयोग का संकेत दिया जाता है। एसएमए वाले अधिकांश रोगियों को घूमने-फिरने के लिए व्हीलचेयर का उपयोग करना पड़ता है।
दुनिया भर के वैज्ञानिक एक ऐसी दवा बनाने के लिए वैज्ञानिक अनुसंधान कर रहे हैं जो एसएमएन प्रोटीन के उत्पादन को बढ़ा सकती है। हालाँकि, फिलहाल, वैज्ञानिक कार्य वांछित परिणाम नहीं लाए हैं।
इस बीमारी की रोकथाम के कोई विशेष तरीके नहीं हैं। गर्भावस्था की योजना के चरण में केवल आनुवंशिकीविद् से परामर्श करने से अजन्मे बच्चे में विकृति विकसित होने का खतरा कम हो जाएगा। यदि आवश्यक हो, तो माता-पिता में पैथोलॉजिकल जीन की उपस्थिति निर्धारित करने के लिए एक आनुवंशिक अध्ययन किया जाता है।
0रोग की आवृत्ति
एसएमए सबसे आम अनाथ (दुर्लभ) बीमारियों में से एक है, जो 6000-10000 में से एक नवजात को प्रभावित करती है।
एसएमए का कारण
एसएमए एक वंशानुगत बीमारी है जो एसएमएन1 जीन में उत्परिवर्तन से जुड़ी है।
रोग प्रकट होने के लिए, माता-पिता दोनों को इस जीन में उत्परिवर्तन का वाहक होना चाहिए। अप्रभावी एसएमए जीन लगभग 40 में से एक है। दो वाहकों से बीमार बच्चे के होने की संभावना 25% है, समान संभावना के साथ दो वाहकों के बच्चे में जीन टूटना नहीं होगा। अन्य 50% मामलों में, वह एसएमए का वाहक होगा, लेकिन वह बीमार नहीं पड़ेगा।
दुर्लभ मामलों में (2% से कम), प्रभावित बच्चे उन परिवारों में पैदा होते हैं जहां केवल एक माता-पिता ही वाहक होते हैं। दूसरे माता-पिता में, एक जीन उत्परिवर्तन तब होता है जब एक अंडा या शुक्राणु रखा जाता है।
उत्परिवर्तन से क्या हानि होती है?
शरीर में एक दोषपूर्ण जीन के कारण मोटर न्यूरॉन्स के जीवित रहने वाले प्रोटीन एसएमएन प्रोटीन का उत्पादन बाधित हो जाता है। इस प्रोटीन के बिना, मोटर न्यूरॉन्स - रीढ़ की हड्डी की तंत्रिका कोशिकाएं जो आंदोलनों और मांसपेशियों की टोन के समन्वय के लिए जिम्मेदार होती हैं - मर जाती हैं, पैरों, पीठ और आंशिक रूप से बाहों की मांसपेशियों को संकेत नहीं मिलता है।
आवश्यक टोन के बिना, मांसपेशियां धीरे-धीरे क्षीण हो जाती हैं। पेट और पीठ की मांसपेशियों की कमी से, अन्य बातों के अलावा, रीढ़ की हड्डी में व्यापक वक्रता आती है, और इससे सांस लेने में समस्या होती है, जो कमजोर मांसपेशियों के कारण पहले से ही होती है।
यह रोग जीवन के पहले महीनों से या बाद की उम्र में प्रकट हो सकता है।

रोग की गंभीरता क्या निर्धारित करती है
एसएमएन प्रोटीन के उत्पादन के लिए दो जीन जिम्मेदार हैं - एसएमएन1 और एसएमएन2।
साथ ही, SMN1 इस प्रोटीन का मुख्य "ग्राहक" है, और SMN2 एक अतिरिक्त है, यह शरीर के सामान्य कामकाज के लिए अपर्याप्त मात्रा में प्रोटीन का उत्पादन करता है। ऐसे मामलों में जहां एसएनएम1 मानव जीनोम में अनुपस्थित है, एसएनएम2 प्रतिस्थापन कार्य करना शुरू कर देता है, लेकिन कभी भी अंतर को पूरी तरह से नहीं भर सकता है।
जीनोम में SMN2 की अधिकतम आठ प्रतियां हैं। रोगी की स्थिति की गंभीरता किसी व्यक्ति के पास मौजूद SMN2 की प्रतियों की संख्या पर निर्भर करती है। रोग का ऐसा जटिल तंत्र इस तथ्य की ओर ले जाता है कि एसएमए के कई रूप होते हैं, और रोगियों की स्थिति बहुत भिन्न होती है।
एसएमए के रूप क्या हैं?
एसएमए के 4 प्रकार होते हैं, जो गंभीरता और बीमारी के पहली बार प्रकट होने की उम्र में भिन्न होते हैं।
एसएमए I, वेर्डनिग-हॉफमैन रोग।बीमारी का सबसे गंभीर रूप 0 से 6 महीने की उम्र के शिशुओं में होता है। इस रूप वाले बच्चों को जन्म से ही सांस लेने, चूसने और निगलने में कठिनाई होती है, और वे सबसे सरल नियंत्रित गतिविधियों में भी महारत हासिल नहीं कर पाते हैं - वे अपना सिर नहीं पकड़ते हैं, वे अपने आप नहीं बैठते हैं। पहले यह सोचा गया था कि बहुसंख्यक (80%) दो वर्ष से अधिक जीवित नहीं रहते थे। अब, नई वेंटिलेशन रणनीतियों और ट्यूब फीडिंग की बदौलत, जीवन प्रत्याशा को कुछ और महीनों तक बढ़ाया जा सकता है।
एसएमए II, डुबोविट्ज़ रोग।रोग की पहली अभिव्यक्तियाँ 7-18 महीनों में होती हैं। इस प्रकार के एसएमए वाला व्यक्ति खा सकता है और बैठ सकता है, लेकिन स्वतंत्र रूप से नहीं चल सकता है। जीवन प्रत्याशा श्वास प्रदान करने वाली मांसपेशियों को क्षति की मात्रा पर निर्भर करती है।
एसएमए III, कुगेलबर्ग-वेलैंडर रोग.यह रोग पहली बार डेढ़ वर्ष के बाद प्रकट होता है। ऐसे मरीज दर्द में खड़े तो हो सकते हैं, लेकिन चल नहीं सकते। एसएमए प्रकार III, एक नियम के रूप में, जीवन प्रत्याशा को प्रभावित नहीं करता है, लेकिन इसकी गुणवत्ता को काफी कम कर देता है।
एसएमएचतुर्थ, इस प्रकार को "वयस्क एसएमए" भी कहा जाता है।चूँकि यह रोग आमतौर पर 35 वर्ष की आयु के बाद प्रकट होता है।
लक्षण मांसपेशियों में कमजोरी, स्कोलियोसिस और कंपकंपी हैं। इसके अलावा, जोड़ों में सिकुड़न (जोड़ों में गतिशीलता की कमी) और चयापचय संबंधी विकार विकसित होते हैं।
रोग की प्रगति बहुत तेज़ नहीं है, मांसपेशियों की कमजोरी पहले पैरों की मांसपेशियों को प्रभावित करती है, फिर बाहों को। आमतौर पर, रोगियों को निगलने और श्वसन क्रिया में कोई समस्या नहीं होती है।
टाइप IV एसएमए वाले अधिकांश मरीज़ चल सकते हैं, और केवल कुछ को ही व्हीलचेयर का सहारा लेना पड़ता है।
एसएमएन जीन के उल्लंघन से जुड़े एसएमए को चिकित्सा साहित्य में समीपस्थ एसएमए कहा जाता है - वे सभी स्पाइनल एमियोट्रॉफी का 95% हिस्सा हैं। एसएमए जो एसएमएन जीन से जुड़े नहीं हैं, वे काफी संख्या में हैं, लेकिन वे दुर्लभ हैं। इनमें शामिल हैं, उदाहरण के लिए, कैनेडी की बीमारी। 1990 के दशक में शोध से पता चला कि कैनेडी की बीमारी एसएमएन1 जीन के टूटने से संबंधित नहीं है, बल्कि अन्य आनुवंशिक उत्परिवर्तन से है जो एसएमएन प्रोटीन के खराब अवशोषण का कारण बनती है। यह रोग 35 वर्ष से अधिक उम्र के लोगों में प्रकट होता है। एसएमपीए की विशेषता मुख्य रूप से अंगों की कमजोरी है।
एक प्रकार का एसएमए जो एसएमएन जीन से जुड़ा नहीं होता है, कहलाता है कैनेडी की बीमारी.इस बीमारी को अभी भी कभी-कभी एसएमए के रूप में जाना जाता है, यह एक पुरानी बात है। 1960 के दशक के उत्तरार्ध में, जब इस शोष का विस्तार से वर्णन किया गया था, तो इसे एसएमए का एक प्रकार माना जाता था, क्योंकि यह तीन प्रकार के एसएमए के समान तंत्रिकाओं और मांसपेशियों को प्रभावित करता है (लेकिन बहुत कम डिग्री तक)।
इसका इलाज कैसे किया जाता है?
आज तक, एसएमए का कोई इलाज नहीं है।
अंतर्राष्ट्रीय निगम "बायोजेन" ने "स्पिनराज़ा" दवा विकसित की है, जिससे उन रोगियों की स्थिति में काफी सुधार हुआ है जिन पर परीक्षण के दौरान इसका उपयोग किया गया था। वर्तमान में, दवा को संयुक्त राज्य अमेरिका में उपयोग के लिए अनुमोदित किया गया है, यूरोप में, कंपनी की गणना के अनुसार, वार्षिक पाठ्यक्रम की अनुमानित लागत लगभग 270 हजार यूरो होगी, रूस में दवा प्रमाणित नहीं है। आजीवन उपचार.
क्या एसएमए से पीड़ित लोगों की मदद करना संभव है और वास्तव में कैसे?
बीमारी का इलाज करना अभी तक संभव नहीं है, लेकिन एसएमए के रोगियों की स्थिति को कम करना संभव है, यानी विभिन्न तरीकों से रोग की अभिव्यक्तियों की भरपाई करना संभव है।
गंभीर प्रकार के एसएमए में, लोगों को सांस लेने और निगलने में मदद की आवश्यकता होती है। इसलिए, उन्हें मोबाइल वेंटिलेटर, एस्पिरेटर, एक्सपेक्टरेंट, अंबू बैग की बेहद जरूरत है।
यहां तक कि एसएमए से पीड़ित बच्चों को भी वास्तव में स्वयंसेवकों की मदद की ज़रूरत होती है जो कम से कम थोड़े समय के लिए अपने माता-पिता की जगह ले सकें।
एसएमए से पीड़ित बच्चों को किसी भी समय मदद की आवश्यकता हो सकती है, इसलिए माता-पिता हमेशा सतर्क रहते हैं और बच्चे के अचानक सांस लेने बंद होने की स्थिति में आवश्यक पुनर्जीवन कौशल सीखते हैं।
कम गंभीर रूप से बीमार रोगियों को सांस लेने में आसानी के लिए दवाओं, कोर्सेट, व्हीलचेयर और अन्य उपकरणों की आवश्यकता होती है जो कमजोर मांसपेशियों वाले लोगों के चलने और जीवन को सुविधाजनक बनाते हैं।
कई वर्षों तक चलने वाली बीमारी थका देने वाली होती है, इसलिए रोगियों, विशेषकर वयस्कों को अक्सर मनोवैज्ञानिक की मदद की आवश्यकता होती है।

यह फंड पूरे रूस में संचालित होता है। फाउंडेशन के काम की दो मुख्य दिशाएँ हैं - एसएमए रोगियों को स्वयं और उनके प्रियजनों को सहायता प्रदान करना, और रूस में एसएमए की स्थिति में प्रणालीगत बदलाव पर काम करना।
आप अपने लिए सुविधाजनक किसी भी तरीके से दान देकर फाउंडेशन की गतिविधियों का समर्थन कर सकते हैं। आप फाउंडेशन के एक विशेष पेज पर एकमुश्त या नियमित दान देकर या एसएमए शब्द और एक स्थान से अलग करके दान की राशि के साथ छोटे नंबर 3443 पर एक एसएमएस भेजकर मदद कर सकते हैं - उदाहरण के लिए, सीएमए 300.
क्या आपको टीकों से एसएमए हो सकता है?
यूरोप और संयुक्त राज्य अमेरिका में, टीकाकरण और बीमारी की अभिव्यक्ति के बीच संबंध का पता नहीं लगाया गया है।
यह समझने के लिए कि क्या एसएमए और टीकाकरण के बीच कोई संबंध है, एसएमए और पोलियो के बीच अंतर को समझा जा सकता है। पोलियोमाइलाइटिस एक संक्रामक रोग है जब प्रारंभिक रूप से स्वस्थ बच्चे का शरीर किसी संक्रमण से क्षतिग्रस्त हो जाता है। क्षतिग्रस्त जीनोम के साथ पैदा हुआ एसएमए वाला बच्चा बाहर से स्वस्थ दिख सकता है, लेकिन वास्तव में वह पहले से ही बीमार है, बस उसकी बीमारी के लक्षण धीरे-धीरे दिखाई देते हैं। इस संबंध में, एसएमए वही "विलंबित" बीमारी है, उदाहरण के लिए, डचेन मस्कुलर डिस्ट्रॉफी या रेट सिंड्रोम, जब एक बच्चा जो कुछ समय के लिए मानक के अनुसार विकास कर रहा है वह पहले से अर्जित कौशल खो देता है और विकलांग हो जाता है।
एसएमए की अधिकांश अभिव्यक्तियाँ पहले मोटर कौशल के विकास से जुड़ी हैं। रोग की पहली अभिव्यक्तियाँ कई आयु-संबंधित टीकाकरणों के साथ समय पर मेल खाती हैं। परिणामस्वरूप, एक व्यक्ति और उसके रिश्तेदार यह दावा कर सकते हैं कि वह "वैक्सीन से बीमार हो गया", लेकिन वास्तव में उसने केवल उस बीमारी के लक्षण दिखाए जो उसे पहले से ही थी।
यह कैसे निर्धारित किया जाता है कि बच्चे को एसएमए है और कोई अन्य बीमारी नहीं?
इस तथ्य के बावजूद कि एसएमए का वर्णन पहली बार 1890 के दशक की शुरुआत में ऑस्ट्रियाई न्यूरोलॉजिस्ट गुइडो वेर्डनिग और जर्मन न्यूरोलॉजिस्ट जोहान हॉफमैन द्वारा किया गया था, बीमारी की प्रकृति पूरी तरह से 20 वीं शताब्दी के अंत में ही समझी गई थी। SMN1 जीन की खोज 1995 में हुई थी। एसएमए के निदान की पुष्टि के लिए आनुवंशिक परीक्षण की आवश्यकता होती है।
रूस में, 2000 के दशक की शुरुआत में उपयुक्त आनुवंशिक परीक्षण उपलब्ध हो गए। अनिवार्य स्वास्थ्य बीमा के अनुसार एसएमए के लिए आनुवंशिक परीक्षण करना संभव है, लेकिन व्यवहार में, बहुत से डॉक्टर इस दुर्लभ निदान को नहीं जानते हैं और रोगियों को उचित अध्ययन के लिए संदर्भित नहीं करते हैं। मॉस्को में वाणिज्यिक प्रयोगशालाओं में ऐसे परीक्षण की लागत लगभग 6 हजार रूबल है।
विशिष्ट निदान की कमी के कारण भी निदान में भ्रम पैदा हुआ है। रूस में एसएमए वाले अधिकांश रोगियों की पहचान नहीं की गई है, पहचाने गए लोगों में से कई को वेर्डनिग-हॉफमैन रोग का निदान किया गया है, हालांकि उनमें से सभी (विशेष रूप से वयस्कों) को वास्तव में इस प्रकार की बीमारी नहीं है।
रूस में एसएमए के कितने मरीज़ हैं?

दवा "स्पिनराज़ा", जो रोगियों की स्थिति में काफी सुधार करती है। फोटो हेल्थबीट.स्पेक्ट्रमहेल्थ.ओआरजी से
रोग की आवृत्ति को ध्यान में रखते हुए, रूस में एसएमए के रोगियों की संख्या सात से चौबीस हजार लोगों तक होनी चाहिए। आज तक, एसएमए फैमिलीज़ फ़ाउंडेशन के मरीज़ों के रजिस्टर में लगभग 400 लोग हैं।
जो रूस में एसएमए से पीड़ित लोगों और उनके परिवारों की मदद करता है
वेरा चैरिटेबल फाउंडेशन, लाइटहाउस के साथ चिल्ड्रेन हॉस्पिस हाउस, चिल्ड्रन पैलिएटिव चैरिटेबल फाउंडेशन, एसएमए फैमिलीज चैरिटेबल फाउंडेशन, मर्सी चिल्ड्रेन पैलिएटिव सर्विस।
2014 से, सेवा "मर्सी" और "एसएमए फैमिलीज़" फाउंडेशन "एसएमए क्लीनिक" की एक संयुक्त परियोजना मास्को में विकसित हो रही है। महीने में एक बार होने वाली बैठकों में, मरीज़ पल्मोनोलॉजिस्ट, आर्थोपेडिस्ट, फिजियोथेरेपिस्ट और मनोवैज्ञानिक से सलाह ले सकते हैं। हाल ही में, कुछ बैठकें वयस्क रोगियों की जरूरतों पर केंद्रित हैं।
एसएमए वाले प्रसिद्ध लोग

इटालियन सिमोन स्पिनोग्लियोएक वंशानुगत बीमारी - स्पाइनल मस्कुलर एट्रोफी टाइप 2 के साथ पैदा हुआ था। वह जन्म से ही चलने में असमर्थ है और केवल इलेक्ट्रिक व्हीलचेयर की मदद से चलती है। लेकिन उसका जीवन पूर्ण और समृद्ध है; उसकी जीने की इच्छा को कोई नहीं रोक सकता।
सिमोना एसएमए हॉटलाइन के इटालियन फैमिलीज के लिए काम करती है और एसएमए और अन्य न्यूरोमस्कुलर रोगों से पीड़ित बच्चों और वयस्कों की मदद करती है।
सिमोन ने इतालवी एसएमए समुदाय में लोकप्रिय कई गाने भी रिकॉर्ड किए - बीमारी के बावजूद आप जो चाहते हैं उसे करने की स्वतंत्रता के बारे में।
रूसी गायिका यूलिया समोइलोवाउख्ता (कोमी गणराज्य) में जन्मी दस साल की उम्र में उन्होंने एक चैरिटी कॉन्सर्ट में प्रस्तुति दी, जिसके बाद उन्हें पायनियर्स के स्थानीय पैलेस में गाने के लिए आमंत्रित किया गया। पंद्रह साल की उम्र में, उन्होंने सिटी हाउस ऑफ़ कल्चर में अध्ययन करना शुरू किया।
2008 में, उन्होंने अपना खुद का संगीत समूह इकट्ठा किया (2010 में भंग कर दिया गया)। 2013 में, उन्होंने रोसिया टीवी चैनल पर फैक्टर ए प्रतियोगिता में भाग लिया। उन्होंने दूसरा स्थान प्राप्त किया और अल्ला पुगाचेवा का व्यक्तिगत पुरस्कार "अल्ला का गोल्डन स्टार" प्राप्त किया। 2017 में, प्रतियोगिता कार्यक्रम में रूस को प्रवेश न मिलने के कारण, वह यूरोविज़न सांग प्रतियोगिता में भाग लेने में असमर्थ रही। व्हीलचेयर में चलता है.
व्लादिमीर वालेरी स्पिरिडोनोव से प्रोग्रामर. उन्होंने स्कूल से स्वर्ण पदक के साथ स्नातक की उपाधि प्राप्त की, फिर अपनी इंजीनियरिंग की डिग्री का बचाव किया। 2015 में, वैलेरी ने मानव सिर के प्रत्यारोपण के लिए इतालवी सर्जन सर्जियो कैनावेरो के प्रयोग में भागीदार बनने की योजना बनाई (प्रयोग रद्द कर दिया गया)।
आज वैलेरी व्लादिमीर के सिटी पब्लिक चैंबर के सदस्य हैं, सुलभ पर्यावरण मुद्दों के विशेषज्ञ हैं, और अपने स्वयं के समुदाय "डिज़ायर फ़ॉर लाइफ" के निर्माता भी हैं, जो एक सुलभ वातावरण बनाने और आशाजनक चिकित्सा परियोजनाओं के बारे में बात करता है। वैलेरी रूसी और विदेशी टीवी पर कई टीवी कार्यक्रमों में भागीदार है।