אמיוטרופיה של עמוד השדרה Werdnig-Hoffmann היא מחלה תורשתית. על רקע הפתולוגיה, נזק בנוירונים מוטוריים מצוין. אמיוטרופיה של עמוד השדרה של הופמן מועברת עם כרומוזומים שאינם מין. לאחר מכן, נבחן מקרוב את המחלה, תמונתה הקלינית ואמצעים טיפוליים אפשריים להעלמתה.
טרמינולוגיה
לפני שנדבר על איך באמיוטרופיה של עמוד השדרה מתבטאת, בואו נכיר כמה מושגים. בואו ננתח את שם הפתולוגיה. הוא מורכב משני חלקים:
- עמוד השדרה - המילה מציינת את לוקליזציה של ההפרה. במקרה זה, אנו מדברים על אלמנט מסוים הממוקם בעמוד השדרה. זהו אחד המבנים החשובים ביותר של הגוף - חוט השדרה.
- אמיוטרופיה היא מילה הכוללת שלושה חלקים: "א" - הפרעות, "מיו" - שריר" ו"גביע" - תזונה.
בהתבסס על מידע זה, ניתן להבין את המשמעות של שם הפתולוגיה. אמיוטרופיה של עמוד השדרה Werdnig-Hoffmann היא אם כן תת תזונה בשרירים. פתולוגיה מאופיינת בנוכחות חולשה ועוויתות של הסיבים.
יְרוּשָׁה
אמיוטרופיה של שרירי עמוד השדרה היא מחלה אוטוזומלית רצסיבית. הגדרה זו מציינת את סוג ההורשה שבה העברת תכונה מתבצעת דרך כרומוזומים שאינם מין. יתר על כן, זה בא לידי ביטוי רק כאשר הוא קיים בתחילה אצל שני ההורים (הם עצמם עלולים לא לחלות).

התפתחות המחלה
אמיוטרופיה של עמוד השדרה אצל מבוגרים אינה מתרחשת. הפתולוגיה מתבטאת בילדים. המחלה מאופיינת במהלך ממאיר ובהתקדמות מהירה. תאים גדולים בחוט השדרה אחראים על תיאום תנועות. הם גם תומכים בטונוס השרירים. כאשר הם ניזוקים, מתפתחת תפקוד לקוי של השרירים.
צורה מולדת
לאמיוטרופיה של עמוד השדרה שלוש צורות. הם נקבעים בהתאם לזמן הביטוי של הסימנים הראשונים ולעוצמת התפתחות התהליך. הצורה המולדת יכולה להתחיל אפילו בתקופה שלפני הלידה. במקרה זה, יש היחלשות של תנועת העובר בשלבים מאוחרים יותר של ההריון. יחד עם זאת, בתחילת התקופה שלפני הלידה, התנועות היו בטווח התקין. עצם הפתרון של ההריון עשוי להיות פתולוגי. לעתים קרובות, כבר בימים הראשונים לאחר הלידה, מתגלה פארזיס שריר בולט, המלווה בירידה בטונוס שלו והחמרה ברפלקסים בגידים. תסמינים רטרובולבריים (מוקדמים) עשויים להופיע גם הם. הם מתבטאים בבכי חלש של תינוק ויניקה איטית. במקרים מסוימים, נצפית ארפלקסיה מלאה. לילד עלולים להיות פרפורים בלשון, היפומיה וירידה ברפלקס הבליעה. אמיוטרופיה של עמוד השדרה מלווה בטכיקרדיה. לעתים קרובות, פתולוגיה משולבת עם מספר מומים, מאט את היווצרות הנפש. אמיוטרופיה של עמוד השדרה מאופיינת במהלך מהיר ומסתיימת בתוצאה קטלנית של 1-1.5 שנים.

צורה מוקדמת
זה מאופיין במהלך מתון יותר מאשר מולד. צורת הילדות המוקדמת נחשבת לביטוי קלאסי של המחלה. אמיוטרופיה של עמוד השדרה במקרה זה מתבטאת בגיל שנה וחצי.
כמעט בכל המקרים, סימני המחלה מתגלים לאחר הרעלת מזון או סוג של נגע זיהומי. ילד שמתפתח בדרך כלל מתחיל לאבד במהירות יכולות מוטוריות שנרכשו בעבר. הוא מפסיק לשבת, לעמוד וללכת. ראשית, פרזיס רפוי בגפיים התחתונות הוא ציין, עובר בהדרגה אל תא המטען והזרועות. מצבו של הילד מתדרדר מהר מאוד. חולשה מופיעה בשרירי הצוואר והשרירים הבולבריים. כתוצאה מאי ספיקה של מערכת הנשימה, דלקת ריאות מופיעה עד גיל 4-5, ואז מתרחש מוות. פרזיס רפוי בילדים מסובך על ידי התכווצויות גידים. לעתים קרובות אמיוטרופיה של עמוד השדרה Werdnig-Hoffman מלווה בהזעת יתר כללית.
התחלה מאוחרת של הפתולוגיה
הצורה השלישית של המחלה מתחילה לאחר 1.5-2 שנים. בהשוואה לקודמים, הוא זורם בקלות יחסית. יכולת התנועה נשארת בילדים עד 10 שנים. לאחר מכן, המצב בדרך כלל מחמיר.

תמונה קלינית
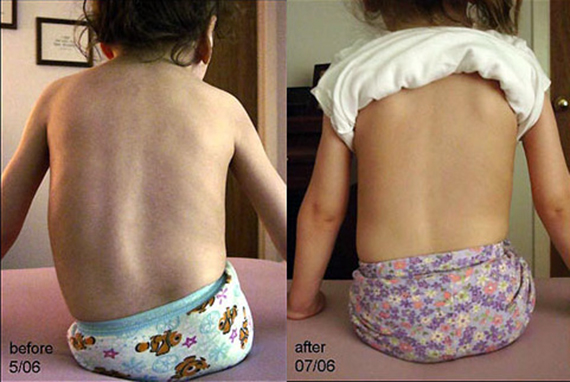
הפתולוגיה מאופיינת בפארזיס, תחילה של הגפיים התחתונות הפרוקסימליות, ולאחר מכן של העליונות. באמיוטרופיה של עמוד השדרה, השכבה התת עורית השומנית באה לידי ביטוי היטב. זה, בתורו, מקשה על זיהוי תפקוד לקוי של השרירים. רפלקסים בגידים מתחילים לדעוך מוקדם מספיק. עבור פתולוגיה, רעד קטן של האצבעות עם זרועות מושטות הוא אופייני. עיוותים בעצמות נחשבים אופייניים, במיוחד בגפיים התחתונות ובעצם החזה. תסמינים בולבריים מתבטאים כאטרופיה של שרירי הלשון עם עוויתות מסוג fibrillar, paresis בחך הרך ורפלקס הלוע מופחת.
מחלת פאציו-לונדון
זוהי גרסה מיוחדת של הביטוי של ניוון. הפתולוגיה מתחילה להתפתח, ככלל, עד גיל שלוש, ובמקרים מסוימים במהלך גיל ההתבגרות. המחלה מאופיינת בחולשה של שרירי הפנים, לרבות שרירי הלעיסה. מציינים קושי בבליעה ושינויים בקול. הפתולוגיה מלווה באטרופיה של הלשון, במקרים מסוימים עלולה להופיע אופטלמופלגיה. המחלה מתקדמת מהר מאוד. לאחר 6-12 חודשים מתרחש מוות. להפרעות בולבריות ניתן להוסיף שיתוק ופרזיס בגפיים. במקרים מסוימים, לתסמינים אלו אין אפילו זמן להתפתח. עם זאת, נתיחה שלאחר המוות מגלה תמיד נגע בתאים של קרני עמוד השדרה הקדמיות לאורך כל הדרך.

אבחון
במהלך הבדיקה, הפתולוגיה מופרדת מהמיוטוניה של אופנהיים. רוב המומחים מאמינים שפתולוגיה זו אינה יחידה נוזולוגית עצמאית. Myotonia Oppenheim, על פי החוקרים, היא תסמונת שבה היפוטוניה של השרירים מסוג בולט הופכת לביטוי המוביל. בהקשר זה, לאחרונה נעשה שימוש נרחב במונח "ילד עצל".
שיטות מחקר: אלקטרומיוגרפיה
זיהוי האמיוטרופיה של עמוד השדרה מבוסס (למעט ביטוי מוקדם ותמונה קלינית טיפוסית) על תוצאות של מספר מחקרים נוספים. מבין אלה, כדאי להדגיש את האלקטרומיוגרפיה. כמעט בכל המקרים, פעילות ספונטנית ביו-חשמלית מזוהה במנוחה בנוכחות פוטנציאל קסם. על רקע התכווצויות רצוניות, מתבררת פעילות חשמלית בעלת אופי מואט עם קצב "פליסדה". הדבר מעיד על עלייה במשך הפוטנציאל ובתופעת הסנכרון.

מחקר פתולוגי
זה מאפשר לך לזהות ירידה במספר התאים של קרני עמוד השדרה הקדמיות, כמו גם שינויים בסוג הניווני. הפרעות פתולוגיות בולטות באזור העיבוי הצווארי והמותני, בגרעינים המוטוריים של עצבי הגולגולת. קיימים גם שינויים בשורשים הקדמיים, באזורים התוך-שריריים של קצות העצבים. יש היעלמות והסתעפות מוגזמת של הטרמינל הרגיל.
ניתוח ביוכימי
מחקר זה מאפשר לך לזהות שינויים במטבוליזם של פחמימות. כך, נמצא כי באמיוטרופיה של עמוד השדרה, הגליקוליזה בחולים קרובה לסוג העובר. לעיתים קרובות מתגלים שינויים משמעותיים בחילוף החומרים של קריאטין-קריאטינין - עליה בהפרשת קריאטינין, ירידה בשחרור קריאטינין. כמו כן, יש לציין כי ריכוז האנזימים בסרום הדם כמעט ללא שינוי.

אמיוטרופיה של עמוד השדרה: טיפול
טיפול פתולוגי מצטמצם למינוי של טיפול בפעילות גופנית ועיסוי. הליכים אלה חייבים להתבצע באופן קבוע. שיטות טיפול רדיקליות נעדרות. במידה מסוימת, נטילת מספר תרופות יכולה להביא להקלה. בפרט, מומחים ממליצים על אמצעים כמו "Sangvinarin", "Galantamine", "Oksazil", "Prozerin". בנוסף, נקבעים ויטמינים מקבוצה B. עם ביטויים חמורים של המחלה, ניתן להמליץ על עירוי דם חוזר במינונים קטנים.
ניוון שרירי עמוד השדרה של ורדניג הופמן היא מחלה נדירה המופיעה במערכת העצבית-שרירית האנושית. מחלה זו מופיעה עקב מוטציות שונות. רופאים מאמינים שהגורם העיקרי לאמיוטרופיה של עמוד השדרה הוא תורשה לקויה.
תסמיני המחלה ישתנו באופן דרמטי בהתאם לחומרת המחלה. רופאים מחלקים באופן מותנה ניוון שרירי עמוד השדרה ל-4 דרגות חומרה. שימו לב שלכל 4 הסוגים יש תכונה משותפת – המטופל חווה כשלים בהתפתחות הנפשית. בכל שלב אינו יכול לגרום לתפקוד לקוי של איברים שונים הממוקמים באזור האגן.
על ניוון של שרירי הידיים, הרגליים והגוף.
הגורם העיקרי למחלה נעוץ בגנטיקה לקויה. מחקרים מדעיים רבים הוכיחו כי ניוון עמוד השדרה מופיע עקב מוטציה בכרומוזום האנושי החמישי. הגן המשתנה אחראי לסינתזה של חלבון מיוחד האחראי להתפתחות תקינה של נוירונים מוטוריים. בשל העובדה כי נוירונים אלה אינם יכולים לבצע באופן מלא את כל תפקידיהם, שרירי הגוף מתחילים להתנוון בבני אדם.
הרופאים אומרים שהמחלה יכולה להופיע בילד רק אם הגן הלא נכון קיים גם אצל האב וגם אצל האם. בנוסף, הרופאים אומרים שכל אדם שני על פני כדור הארץ הוא נשא של הגן הלא נכון.
תסמינים בצורה הראשונה והשנייה של המחלה
אמיוטרופיה בעמוד השדרה של Werdnig-Hoffmann סוג 1 מופיעה לרוב בששת החודשים הראשונים לחייו של ילד. במקרים מסוימים ניתן לזהות את המחלה במהלך האבחון במהלך הלידה. בשלב 1, לילד יש טונוס מופחת של סיבי שריר, ורפלקסים בגידים עשויים להיעדר לחלוטין. לפעמים תפקוד הבליעה סובל. הלשון, ככלל, גם מתנוונת. בשלב 1, תפקוד העיכול של הילד מופרע. בגלל זה, מזון יכול להיכנס לדרכי הנשימה. גם הנשימה מופרעת, בגלל זה, עלולה להופיע אי ספיקת ריאות. כמו כן, בילדים עם אמיוטרופיה בעמוד השדרה מדרגה 1, התפתחות המיומנויות המוטוריות נפגעת ונצפה עיוות בחזה. על פי הסטטיסטיקה, אם המחלה מתחילה להתקדם מיד לאחר הלידה, הילד מת ב-3-4 החודשים הראשונים לחייו.
עם ניוון עמוד השדרה מדרגה 2, הילד מפתח תסמינים מדאיגים רק 6 חודשים לאחר הלידה. בתחילה, המטופל מפתח ניוון של שרירי הירך. רפלקסים בגידים מתעממים בהדרגה. שרירי הפנים, ככלל, אינם נתונים לאטרופיה. במקרים מסוימים, למטופל יש רעידות בידיים והנשימה מופרעת. גם שרירי הצוואר נחלשים עם הזמן. עם הזמן, החולה מפתח עקמת או נקע של מפרק הירך. ילדים עם ניוון בעמוד השדרה דרגה 2 רגישים יותר לזיהומים פתוגניים עקב פגיעה בתפקוד הנשימה. במקרה של טיפול בטרם עת, החולה עלול למות מחנק.
: גורמים ושיטות טיפול במחלה.
מה לעשות אם מושכים את הגב התחתון? למידע נוסף על טיפולים.
איך עושים את זה בצורה נכונה ויעילה?
תסמינים בסוג השלישי והרביעי של אמיוטרופיה
אמיוטרופיה של עמוד השדרה של Werdnig-Hoffman שלב 3 מופיעה, ככלל, בגיל ההתבגרות. סימפטום אופייני לסוג זה של מחלה הוא הליכה לא יציבה. זה מוסבר על ידי העובדה כי הטון של שרירי הרגליים מופחת בחדות. זאת בשל העובדה שסיבי השריר נעשים דקים יותר. החולה עלול לעתים קרובות למעוד או אפילו ליפול. עם הזמן, החולה עלול להפסיק ללכת לחלוטין. לעיתים מופיעה ניוון בשרירי הידיים. גם הבעות הפנים של המטופל מופרעות. השלד עובר שינויים. החזה של המטופל מעוות ועבודת המפרקים מופרעת. במקרים נדירים, החולה מופרע על ידי נשימה תקינה.
אמיוטרופיה של עמוד השדרה של Werdnig-Hoffman שלב 4 מופיעה, ככלל, בבגרות. סימפטום אופייני בשלב זה של המחלה הוא התרחשות של חולשה ברגליים. החולה עלול להתלונן על כאבי שרירים חזקים. עם הזמן, שרירי הרגליים מתחילים להתנוון, והרפלקסים נעשים עמומים. התקדמות ניוון שרירי עמוד השדרה מובילה לעובדה שהמטופל מפסיק ללכת. בדרגה 4, תפקוד הנשימה אינו משתנה. שרירי הידיים פועלים במלואם. הרופאים מאמינים שסוג 4 של המחלה הוא שפיר.
אבחון וטיפול בפתולוגיה
אם לאדם יש את הסימפטומים הראשונים של ניוון עמוד השדרה, נקבע לו אבחנה מקיפה. ראשית, על המטופל לעבור בדיקת אלקטרונואורומיוגרפיה. עם הליך זה, רופאים יכולים לזהות חריגות בתפקוד של נוירונים. לאחר מכן, על החולה לעבור מחקר גנטי. במהלך הליך זה, הרופאים חוקרים נתונים המתקבלים מה-DNA של המטופל. מחקר גנטי מגלה מוטציות בכרומוזום 5.
אם לאישה יש קרובי משפחה שחלו במחלה זו, במהלך ההריון עליה לעבור אבחון טרום לידתי מיוחד. אם מתגלה פתולוגיה בעובר, ייתכן שיהיה צורך בהפסקת הריון כפויה.
ניוון עמוד השדרה אינו מטופל. עם זאת, ניתן להקל על מצבו של החולה בעזרת תרופות מיוחדות המשפרות תהליכים מטבוליים ברקמות העצבים. הטיפול מתווסף בויטמיני B, קורס של סטרואידים אנבוליים, טיפול בפעילות גופנית ופיזיותרפיה.
מקורות נוספים:
- 1. Su Y.N., Hung C.C., Lin S.Y., Chen F.Y., Chern J.P., Tsai C. סקר לאטרופיה של שרירי עמוד השדרה (SMA) // הספרייה הציבורית למדע
- 2. Sugarman E. A., Nagan N., Zhu H., Akmaev V. R., Zhou Z., Rohlfs E. M., Flynn K., Hendrickson B. C., Scholl T., Sirko-Osadsa D. A., Allitto B. A. סקר נשאים פאן-אתני ואבחון טרום לידתי לאטרופיה של שרירי עמוד השדרה: ניתוח מעבדה קליני כתב העת האירופי לגנטיקה אנושית.
- 3. אמיוטרופיה של עמוד השדרה מסוג I, II, III, IV... // המרכז לגנטיקה מולקולרית של המרכז למחקר גנטי רפואי.


מכל ניווני שרירי עמוד השדרה הקיימים והידועים למדע אמיוטרופיה של ורדניג-הופמןהוא המגוון הכבד ביותר.
השכיחות של מחלה זו היא כעת בסביבות מקרה אחד לכל 7-11 אלף תינוקות שזה עתה נולדו.
הגן הגורם למחלה זו קיים בכל אדם 50.
עם זאת, עקב תורשה אוטוזומלית רצסיבית, הפרעה מתרחשת בילד רק כאשר לשני ההורים יש מידע גנטי זה.
לכן, ההסתברות שהתינוק ייוולד עם פתולוגיה, במקרה זה, היא כ-25%.
האם ניתן להתמודד עם מחלה כזו, או לפחות לעצור את התקדמות הסימפטומים, נספר במאמר זה.
מהי אמיוטרופיה של ורדניג-הופמן?
אמיוטרופיה של עמוד השדרה סוג 1 או, במילים אחרות, אמיוטרופיה של עמוד השדרה Werdnig-Hoffmann- זוהי מחלה מיוחדת של מערכת העצבים, העוברת בתורשה (לרוב משני ההורים). פתולוגיה זו מאופיינת בנוכחות של חולשת שרירים כמעט בכל מערכת השרירים של הגוף. ילד הסובל ממחלה כזו לא יכול לשבת, לזוז ולשרת את עצמו בכוחות עצמו.
למרבה הצער, אין תרופה לסוג זה של מחלה בעולם. המקסימום שרופאים יכולים להציע בזמננו הוא אבחון טרום לידתי. בדיקה כזו עוזרת למנוע לידת תינוק חולה במשפחה.
הפתולוגיה קיבלה את שמה משני מדענים שתיארו אותה לראשונה בסוף המאה ה-19. נכון לעכשיו, המושג אמיוטרופיה של עמוד השדרה מתייחס למספר צורות של המחלה השונות מבחינה קלינית. אבל כולם קשורים באותו פגם גנטי שיש להורי הילד.
תמונה קלינית של המחלה
לאמיוטרופיה של עמוד השדרה יש כמה צורות וזנים, שכל אחד מהם שונה בגיל הופעת התסמינים האופייניים, חומרת מהלך המחלה ותוחלת החיים של החולים.
בְּדֶרֶך כְּלַל פתולוגיה זו מובילה לנכות, שכן המערכת המוטורית של הגוף מופרעת, והמטופל אינו מסוגל לנוע באופן עצמאי או לשרת את עצמו באופן עצמאי. במצבים קליניים חמורים, ייתכן שיהיה צורך בפיקוח רפואי מתמיד בחיי היומיום.

כסאות גלגלים, הליכונים, קביים, קנים עוזרים למטופל כזה לנוע. מחלה כזו עלולה להוביל לתוצאה קטלנית רק כאשר מופיעים סיבוכים ממערכת הנשימה והלב וכלי הדם (עם דלקת ריאות ואי ספיקת לב).
סיבי עצב רגישים אינם נופלים תחת השפעת הפתולוגיה, כך שהילד שומר על כל מיני רגישות. גם האינטלקט והתפקודים המנטליים אינם סובלים, ולכן בעת הלמידה, הילד קולט ומטמיע מידע באופן די נורמלי.
סיווג מחלות
בהתאם לגיל שבו הופיעו התסמינים האופייניים למחלה, האמיוטרופיה של Werdnig-Hoffman מחולקת למספר סוגים:
- צורה מולדת של פתולוגיה. גיל משוער של הופעת השינויים: מ-0 עד 6 חודשים. מאופיין בדרך כלל בתנועת עובר תוך רחמית חלשה. עם צורה מולדת, תת לחץ דם בשרירים נצפה מהימים הראשונים לחייו של התינוק. תוך זמן קצר, רפלקסים עמוקים דועכים: הילד בוכה חלש, מוצץ חלב אם או פטמה גרוע, לא יכול להחזיק את ראשו. לפעמים קורה שהתסמינים האלה מופיעים קצת מאוחר יותר, אז התינוק יכול ללמוד להחזיק את הראש ולשבת, אבל בגלל שיש הפרה, כישורים אלה לא יתפתחו אצלו. כמו כן, הצורה המולדת עשויה להיות מלווה בהפרעות בולבריות, ירידה ברפלקס הלוע ועוויתות פשקולריות של הלשון. הצורה המולדת נחשבת לממאירה ביותר ולרוב יכולה לשלב גם אוליגופרניה, עיוותים בחזה ו-4 דרגות עקמת. חוסר תנועה מהיר ופרזיס של מערכת הנשימה מוביל לכשל נשימתי ולאחר מכן למוות;
- צורת ילדות מוקדמת.עם סוג זה של פתולוגיה, הסימפטומים הראשונים עשויים להופיע לאחר 6 חודשים. בשלב זה, לילדים יש התפתחות גופנית ונפשית תקינה. הם מתחילים לרכוש לאט לאט את הכישורים הטבעיים הראשונים, כמו היכולת להחזיק את הראש, לעמוד, לשבת ולהתהפך. ברוב המקרים, עם סוג זה של מחלה, ילדים לעולם לא ילמדו ללכת. בשלב הראשוני, paresis מתרחשת בגפיים התחתונות, ואז די מהר הם מתפתחים בגפיים העליונות ובכל השרירים. תת לחץ דם שרירי נכנס, רפלקסים עמוקים מתפוגגים, רעד באצבעות, התכווצויות שרירים לא רצוניות עלולות להופיע. בשלבים מאוחרים יותר מתווספות הפרעות בולבריות ואי ספיקת נשימה (פרוגרסיבית) לכל התסמינים. צורה זו של המחלה ממשיכה לאט יותר מהסוג המולד. מטופלים יכולים לחיות עד 15 שנים;
- אמיוטרופיה של קוגלברג-וולנדר.השפיר ביותר מכל צורות האמיוטרופיה של עמוד השדרה. התסמינים מופיעים לאחר שנתיים, לפעמים בין השנים ה-15 ל-30. עם צורה זו, אין פיגור שכלי בהתפתחות; במשך זמן רב, המטופלים מסוגלים לנוע באופן עצמאי. רבים חיים עד זקנה בשלה בשירות עצמי מלא.
גורמי סיכון וגורמים למחלה
מאז וורדניג-הופמן אמיוטרופיה של עמוד השדרה היא מחלה תורשתית, אם כן הסיבות להתרחשותו נעוצות בקוד הגנטי של שני הוריו של המטופל. הבעיה נעוצה בכרומוזום החמישי, שעובר מוטציה גנטית.
משנה את הגן שאחראי על ייצור החלבון SMN. בגוף בריא, הסינתזה של חלבון זה מבטיחה התפתחות תקינה של נוירונים מוטוריים. אם הוא עבר מוטציה, אז הנוירונים המוטוריים מתחילים להתמוטט, מה שמוביל לשיבוש בהעברת הדחפים מסיב העצב לשריר. כתוצאה מכך, השריר אינו מתפקד. לכן יש ניוון מוטורי וחוסר יכולת לנוע כרגיל.

גן עם הפרעות יש דפוס תורשה אוטוזומלית רצסיבית. המשמעות היא שכדי שהמחלה תתפתח, נדרשת התאמה של שני גנים שעברו מוטציה משני ההורים. הָהֵן. למעשה, גם האב וגם האם חייבים להיות נשאים של הגן עם הפתולוגיה.
יחד עם זאת, הם לא חולים, כי יש להם גן בריא דומיננטי (זה גם בגלל זיווג הגנים). אם גם לאבא וגם לאמו של התינוק יש גן עם פתולוגיה, אז הסיכון שהילד ייוולד עם הפרעות הוא 25%.
סרטון: "מהי ניוון שרירי עמוד השדרה?"
שיטות לאבחון המחלה
בכל הנוגע לאבחון מחלה מסוג זה יש לקחת בחשבון שלנוירולוג שיבצע את הבדיקה יש חשיבות רבה לגיל בו מופיעים התסמינים הראשונים אצל התינוק.
גַם הדינמיקה של התפתחות הסימפטומים חשובה, נתוני מצב נוירולוגי (כלומר, נוכחות / היעדר הפרעות מוטוריות מסוג פריפריאלי על רקע רגישות נורמלית), נוכחות / היעדר חריגות מולדות נוספות ועיוותים בעצמות (עקמת, קיפוזיס, לורדוזיס, טורטיקוליס).
ניתן לזהות את הסוג המולד של המחלה על ידי רופא ילודים. הבדיקה מתבצעת עם מיופתיות, ניוון שרירים (פרוגרסיבי), טרשת צדדית אמיוטרופית, פוליומיאליטיס, שיתוק מוחין, וכו '. אם האבחנה דורשת את האישור המדויק ביותר, אזי נעשה שימוש גם באלקטרו-נוירומיוגרפיה (מחקרים של המנגנון העצבי-שרירי).
 האבחנה הסופית נקבעת רק לאחר קבלת נתונים על הביולוגיה של השרירים ולימוד המצב הגנטי.. מחקר ניתוח ה-DNA מאפשר לגנטיקאים לגלות את הסחף ההטרוזיגוטי של סטיית גנים (חשוב בעת תכנון ההריון הבא). ניתוח כמותי של מספר הגנים של לוקוס SMA מתבצע גם (מאפשר לך לחשב נוכחות של גן פתולוגי אצל ההורים.
האבחנה הסופית נקבעת רק לאחר קבלת נתונים על הביולוגיה של השרירים ולימוד המצב הגנטי.. מחקר ניתוח ה-DNA מאפשר לגנטיקאים לגלות את הסחף ההטרוזיגוטי של סטיית גנים (חשוב בעת תכנון ההריון הבא). ניתוח כמותי של מספר הגנים של לוקוס SMA מתבצע גם (מאפשר לך לחשב נוכחות של גן פתולוגי אצל ההורים.
בדיקת DNA טרום לידתית יכולה לעזור להפחית את הסיכוי ללדת תינוק עם מחלת ורדינג-הופמן. אבל הקושי כאן טמון בעובדה ששיטות פולשניות של אבחון טרום לידתי (ביופסיה כוריונית, קורדוקנטזה, בדיקת מי שפיר) משמשות להשגת חומר DNA.
אם המחלה מאושרת ברחם, זו תהיה אינדיקציה להפסקה מלאכותית של ההריון הנוכחי.
טיפול במחלה
הרפואה המודרנית, למרבה הצער, עדיין לא פיתחה תרופות שיכולות להתמודד עם סוגים שונים של מוטציות גנטיות, ולכן לא קיים קורס טיפול שיכול להביס את האמיוטרופיה של ורדניג-הופמן. ישנן מספר פעילויות שיכולות להאט את ניוון השרירים המתקדם, אך אין לצפות לניסים.
תרופות
תרופות יכול רק להחליש את מצבו של החולה, תומכים במקצת ומזינים את מערכת העצבים שלו, אבל הם לא יכולים להביס את הגורם להתפתחות המחלה.
עם אמיוטרופיה של עמוד השדרה, משתמשים בתרופות הבאות:
- סרברוליזין, ציטופלבין, חומצה גלוטמית, ATP, קרניטין כלוריד, מתיונין, אשלגן אורוטאטואחרות - תרופות אלה משפרות במידת מה את חילוף החומרים של השרירים ורקמות העצבים, ולכן הן נקבעות לצריכת קורס תקופתית;
- ויטמינים מקבוצת B(Neurovitan, Milgamma);
- סטרואידים אנבוליים(Retabolil, Nerobol);
- Prozerin, Neuromidin, Dibazol- תרופות אלו יכולות לשפר במידה מסוימת את מוליכות השרירים.

פעילות גופנית, עיסוי, טיפול בפעילות גופנית
והאם ידעת ש…
עובדה הבאה
תרגילים גופניים ועיסוי משמשים גם כדי להקל על מצבו של המטופל.. עם זאת, במקרה של פתולוגיה כה רצינית, טיפול בתרפיה ועיסוי צריכים להיקבע על ידי הרופא שלך ולהתבצע תחת שמירה קפדנית של מומחה (כך גם לגבי פיזיותרפיה). לא ניתן להשתמש כאן במתחמי טיפול בסיסיים בתרגול, אלא רק בבחירה אישית על ידי הרופא בהתאם לרמת ההתפתחות של הפתולוגיה, לתסמינים האופייניים ולשלב המחלה.
סרטון: "טיפול באטרופיה של שרירי עמוד השדרה באמצעות תרפיה גנטית"
טיפול בבית
טיפול לא מורשה בבית ללא התייעצות ופיקוח רופא אסור בהחלט במקרה זה.. אמצעי מניעה בבית ניתן ליישם רק אם הרופא נתן אישור. מומחה יכול לרשום לך כמה תרגילים פשוטים שאתה יכול לעשות בבית. אם לא היו המלצות כאלה, אז אסור בהחלט לעשות משהו בעצמך.
מניעת מחלות
אם כן, בהתבסס על העובדה שאמיוטרופיה של וורדינג-הופמן היא מחלה תורשתית אין אמצעי מניעה לכך. למרבה הצער, אם הפתולוגיה של התינוק התגלתה אפילו בתקופה שלפני הלידה, יהיה מומלץ לפנות להפסקה מלאכותית של הריון לא מוצלח. טרם הומצאו טיפול או מניעה לסוג זה של מחלה.
פרוגנוזה של מחלה
למרבה הצער, הפרוגנוזה לפתולוגיה זו היא שלילית ביותר.. אין סיכוי להחלמה ולא יכול להיות. אם ילד נולד עם צורה מולדת של אמיוטרופיה, אז הוא ימות בין 6 חודשים לגיל שנתיים. סוגים מאוחרים יותר של המחלה יאריכו את חיי החולה, אך הסבירות למוות נותרה גבוהה מאוד.
סיכום
הרפואה המודרנית מכירה מחלות רבות ששום שיטה ושום תרופה לא יכולים להתמודד איתן. אמיוטרופיה של עמוד השדרה ורדניג-הופמןאחד מהם. המקור הגנטי של הפתולוגיה הזו איפשר לטפל בה בשיטות המוכרות לנו.
יש גם כמה נקודות חשובות יותר שכדאי לשקול אם אתה בסיכון ללדת תינוק עם מחלה זו:
- לאמיוטרופיה של עמוד השדרה Werdnig-Hoffman יש כמה זנים: אמיוטרופיה מולדת, מוקדמת וקוגלברג-וולנדר. הצורה המולדת מביאה למוות מוקדם (בערך בגיל 6 חודשים עד שנתיים), הצורה המוקדמת והמאוחרת יכולה להאריך את חיי החולה, אך אין סיכוי להחלמה כזו או אחרת;
- הסיכון לפתח פתולוגיה יכול להופיע רק אם לשני ההורים יש גן שעבר מוטציה. רק בתנאי שגם האם וגם האב (שאינם נראים חולים, אלא רק נשאים של הגן) הם הבעלים של הפרעות בשרשרת הגנטית, יכול להיוולד ילד עם מחלת ורדניג-הופמן (סיכוי של 25%. );
- אי אפשר לרפא מחלה גנטית, כשם שאי אפשר לבצע מניעה.. תרופות ניתנות רק כדי להקל על הסימפטומים של המטופל ולשמור על הטונוס בגוף. טיפול בפעילות גופנית ועיסוי נקבעים רק על ידי הרופא המטפל, רק על ידו ולא על ידי אף אחד אחר, שכן המחלה חמורה ודורשת השגחה קפדנית. אם האבחנה הטרום לידתית כבר חשפה את העובדה שהילד ייוולד עם פתולוגיה, אז הרופאים מייעצים לפנות להפסקה מלאכותית של הריון.
לעבור את המבחן!
ניוון שרירי עמוד השדרה של ורדניג-הופמן היא פתולוגיה נוירו-שרירית מתקדמת שעוברת בתורשה. המחלה מובילה לפגיעה בשרירים המפוספסים הפרוקסימליים של תא המטען, הגפיים התחתונות והצוואר.
גורם ל
זוהי פתולוגיה תורשתית, אשר מבוססת על מוטציה בכרומוזום 5 אנושי. כתוצאה מכך, הסינתזה של חלבון SMN, הנדרש להתפתחות תקינה של נוירונים מוטוריים, מופרעת.
המחלה מובילה להרס או התפתחות של תאי עצב פגומים שאינם מסוגלים להעביר דחפים לסיבי השריר. לכן, השרירים המועצבים מפסיקים לעבוד, ניוון מתפתח.
על פי הסטטיסטיקה, כל אדם שני הוא נשא של גן פתולוגי.
לגן שעבר מוטציה יש דפוס תורשה אוטוזומלית רצסיבית- להתפתחות המחלה נדרשת התאמה של שני כרומוזומים שגויים מהאם והאב.

ילד חולה נולד רק מהורים הנושאים את הגן הפתולוגי. עם זאת, לאם ולאב יש גן דומיננטי בריא, ולכן הם אינם מראים תסמינים של פתולוגיה.
תמונה קלינית של המחלה
הסימפטומולוגיה וחומרת מהלך ה-SMA נקבעת לפי זמן הביטוי של הסימנים הראשונים של המחלה. לכן, הרופאים מבחינים בין 3 סוגי מחלות:
- צורה מולדת;
- ילדות מוקדמת;
- צורה מאוחרת.

כדאי לשקול ביתר פירוט כל אחת מהטפסים.
תכונות של הצורה המולדת
לידתם של ילדים עם paresis רפוי היא אופיינית. יילודים מאובחנים עם היפוטוניה כללית של השרירים, היעדר רפלקסים עמוקים. הפרעות Bulbar מובילות לעובדה שהתינוק יונק באיטיות את השד, צורח בשקט, רפלקס הלוע שלו מופחת. עם הזמן, הילד מאובחן עם paresis של שריר הסרעפת.
תסמינים של אמיוטרופיה של עמוד השדרה של Werdnig-Hoffman מתרחשים אצל 1 מתוך 10,000 יילודים.
המחלה קשורה לעתים קרובות עם עיוותים בעצמות ובמפרקים(סטרנום משפך, עקמת, התכווצות מפרקים). התפתחות איטית של תפקודים סטטיים ותנועתיים אופיינית, כך שרק מספר מצומצם של תינוקות מסוגלים להחזיק את ראשם בכוחות עצמם. עם זאת, מיומנות זו יכולה לחזור במהירות.

חולים רבים עם צורה מולדת של מיופתיה בעמוד השדרה סובלים מאינטליגנציה מופחתת, מומים (קלת רגליים, הידרוצפלוס, המנגיומה, קריפטורכידיזם).
המחלה מתפתחת במהירות ומאופיינת במהלך ממאיר. חולים חיים רק לעתים רחוקות יותר מגיל 9 שנים. סיבת המוות היא פתולוגיה סומטית.
יותר מ-50% מהילדים הסובלים מצורה מולדת של אמיוטרופיה הופמן-וורדניג אינם חיים עד שנתיים.
תסמינים של צורת הילדות המוקדמת
התסמונת מאופיינת בהתפתחות הסימנים הראשונים לפתולוגיה אצל תינוקות מעל גיל 6 חודשים. יחד עם זאת, למטופלים יש התפתחות מוטורית מספקת: הם מסוגלים להחזיק את הראש, לשבת ולפעמים הם יכולים לעמוד. התפתחות תת-חריפה של המחלה על רקע שיכרון או מחלה זיהומית אופיינית.

מיופתיהמוביל להופעת paresis רפוי של הרגליים, אשר מתפשט במהירות לשרירי תא המטען והזרועות. זה מעורר ירידה בטונוס השרירים, רפלקסים עמוקים. בשלבים המאוחרים יותר, הילד מאובחן עם תת לחץ דם כללי, שיתוק בולברי.
הפרוגנוזה לפתולוגיה היא שלילית - למחלה יש מהלך ממאיר, ילדים חיים רק לעתים רחוקות עד 15 שנים.
צורה מאוחרת
הסימנים הראשונים לפתולוגיה מופיעים לאחר היווצרות מלאה של פונקציות תנועתיות וסטטיות. לכן, ילדים רבים מסוגלים לרוץ וללכת בכוחות עצמם. אופייני הוא התפתחות הדרגתית של מיופתיה, המתבטאת בתנועות מביכות ולא ודאות של הילד. בהדרגה, ההליכה מתחילה להשתנות - הילדים הולכים כמו בובות שעון, מכופפים ללא הרף את הברכיים.

פרזיס רפוי ממוקם בתחילה בגפיים התחתונות, אך מתפשט בהדרגה לשרירי תא המטען והזרועות. לילד יש שומן תת עורי מפותח, כך שהניוון של סיבי השריר בקושי מורגש. בהדרגה, החולה מפתח תסמינים בולבריים, יש רעד של האצבעות.
בשלבים הראשונים של SMA, רפלקסים עמוקים דוהים, והחזה מתחיל להתעוות.
למחלה יש מהלך ממאיר, אך התסמינים מתפתחים לאט יותר מאשר בצורות קודמות. ילדים מאבדים את היכולת לנוע באופן עצמאי רק עד גיל 10. המוות מתרחש בדרך כלל לפני גיל 30.
| סוג SMA | מניפסט מחלה | מקסימום תפקוד | גיל המוות |
| צורה מולדת | התסמינים הראשונים מתפתחים בילדים מתחת לגיל 6 חודשים. | הילד לא מסוגל לזוז, להחזיק את הראש, לשבת | חולים רבים מתים לפני גיל שנתיים אך עשויים לחיות עד גיל 9 |
| צורת ילדות מוקדמת | התסמינים מתחילים בין 7 ל-12 חודשים | המטופל יכול לשבת, לעמוד, אך בהדרגה הפונקציות נסוגות | בני 14-15 |
| צורה מאוחרת | סימני המחלה מתפתחים בילדים מעל גיל שנה | הילד עומד והולך | גיל מ 20 עד 30 שנים |
אמצעי אבחון
לאבחנה יש חשיבות רבה לגיל הסימפטומים הראשונים של הפתולוגיה, הדינמיקה של ההתפתחות והמצב הנוירולוגי של המטופל (הפרעות בתפקודים מוטוריים על רקע שימור הרגישות), נוכחות של עיוות עצם ואנומליות מולדות. הצורה המולדת של SMA מאובחנת בדרך כלל על ידי ניאונטולוגים.
אבחון מקיף כולל את הפעילויות הבאות:

בדיקת DNA טרום לידתי מומלצת כדי להפחית את הסיכון ללדת תינוק עם SMA. עם זאת, ניתן לקבל חומר לאבחון רק באמצעות טכניקות פולשניות (דיקור מי שפיר, ביופסיה כוריונית, קורדוקנטזה). אם אמיוטרופיה אובחנה ברחם, יש לציין הפסקת הריון.

תכונות הטיפול במחלה
מיופתיה בעמוד השדרה היא פתולוגיה חשוכת מרפא, ולכן הטיפול יכול רק להקל על מצבו של המטופל. לשם כך משתמשים בתרופות הבאות:
- סטרואידים אנבוליים;
- ויטמינים מקבוצת B;
- אמצעים המשפרים את חילוף החומרים של השרירים והנוירונים;
- תרופות המשפרות את ההולכה העצבית-שרירית.
בנוסף, נקבע קורס עיסוי וטיפול בפעילות גופנית, פיזיותרפיה (גירוי שרירים חשמלי, טיפול בחמצן), תיקון אורטופדי. על המטופל להקפיד על דיאטה תזונתית.

אם מתפתח כשל נשימתי, הילד החולה מחובר למכונת הנשמה כדי להחזיר את הנשימה. עם הפרה בולטת של רפלקס הבליעה, השימוש בצינור האכלה גסטרוסטומיה מצוין. רוב החולים עם SMA צריכים להשתמש בכיסא גלגלים כדי להתנייד.
מדענים מרחבי העולם עורכים מחקר מדעי ליצירת תרופה שיכולה להגביר את ייצור החלבון SMN. עם זאת, כרגע, עבודה מדעית לא הביאה את התוצאה הרצויה.
למחלה אין שיטות מניעה ספציפיות. רק התייעצות עם גנטיקאי בשלב תכנון ההריון תפחית את הסיכון לפתח פתולוגיות אצל ילד שטרם נולד. במידת הצורך, מתבצע מחקר גנטי לקביעת נוכחות של גן פתולוגי אצל ההורים.
0תדירות מחלות
SMA היא אחת המחלות היתומות (נדירות) הנפוצות ביותר, הפוגעת ביילוד אחד ב-6000-10000.
גורם ל-SMA
SMA היא מחלה תורשתית הקשורה למוטציות בגן SMN1.
כדי שהמחלה תתבטא, שני ההורים חייבים להיות נשאים של המוטציה בגן זה. הגן הרצסיבי של SMA הוא אחד מכל 40 בערך. ההסתברות ללדת ילד חולה משני נשאים היא 25%, באותה הסתברות שלילד משני נשאים לא תהיה התמוטטות גנים. בעוד 50% מהמקרים הוא יהיה נשא של SMA, אך לא יחלה.
במקרים נדירים (פחות מ-2%), ילדים שנפגעו נולדים במשפחות שבהן רק הורה אחד הוא נשא. בהורה השני מתרחשת מוטציה בגן כאשר מוטלת ביצית או זרע.
מה נפגע ממוטציה
עקב גן פגום בגוף, ייצור חלבון SMN, חלבון ההישרדות של נוירונים מוטוריים, מופרע. ללא חלבון זה, נוירונים מוטוריים - תאי העצב של חוט השדרה האחראים על תיאום התנועות וטונוס השרירים - מתים, האות לשרירי הרגליים, הגב, ובחלקו הזרועות לא עובר.
ללא הטון הדרוש, השרירים מתנוונים בהדרגה. המחסור בשרירי הבטן והגב מוביל, בין היתר, לעיקול נרחב של עמוד השדרה, והם מובילים לבעיות נשימה, שכבר קיימות עקב שרירים חלשים.
המחלה יכולה להתבטא כבר מחודשי החיים הראשונים או בגיל מאוחר יותר.

מה קובע את חומרת המחלה
שני גנים אחראים לייצור חלבון SMN - SMN1 ו-SMN2.
יחד עם זאת, SMN1 הוא "הלקוח" העיקרי של חלבון זה, ו-SMN2 הוא אחד נוסף; הוא מייצר חלבון בכמות שאינה מספקת לתפקוד תקין של הגוף. במקרים בהם SNM1 נעדר בגנום האנושי, SNM2 מתחיל לבצע פונקציות החלפה, אך לעולם לא יכול למלא את החסר במלואו.
ישנם עד שמונה עותקים של SMN2 בגנום. חומרת מצבו של המטופל תלויה במספר העותקים של SMN2 שיש לאדם. מנגנון מורכב כזה של המחלה מוביל לעובדה של-SMA יש כמה צורות, ומצב החולים שונה מאוד.
מהן הצורות של SMA?
ישנם 4 סוגים של SMA, הנבדלים זה מזה בחומרתם ובגיל שבו המחלה מופיעה לראשונה.
SMA I, מחלת ורדניג-הופמן.הצורה החמורה ביותר של המחלה מתרחשת אצל תינוקות בין גיל 0 ל-6 חודשים. ילדים עם צורה זו מלידה מתקשים לנשום, לינוק ולבלוע, וגם לא שולטים בתנועות המבוקרות הפשוטות ביותר - הם לא מחזיקים את הראש, הם לא יושבים בכוחות עצמם. בעבר חשבו שהרוב (80%) לא חי מעבר לגיל שנתיים. כעת, הודות לאסטרטגיות אוורור חדשות והזנת צינורית, ניתן להאריך את תוחלת החיים בעוד כמה חודשים.
SMA II, מחלת דובוביץ.הביטויים הראשונים של המחלה בגיל 7-18 חודשים. אדם עם SMA מסוג זה יכול לאכול ולשבת, אך אינו הולך באופן עצמאי. תוחלת החיים תלויה במידת הפגיעה בשרירים המספקים נשימה.
SMA III, מחלת קוגלברג-וולנדר.המחלה מתבטאת לראשונה לאחר שנה וחצי. חולים כאלה יכולים לעמוד (בכאב), אך לא ללכת. SMA סוג III, ככלל, אינו משפיע על תוחלת החיים, אך פוגע מאוד באיכותו.
SMAIV, סוג זה נקרא גם "SMA למבוגרים",מאחר והמחלה מתבטאת בדרך כלל לאחר גיל 35 שנים.
התסמינים הם חולשת שרירים, עקמת ורעד. בנוסף, מתפתחים התכווצויות מפרקים (הגבלה בתנועתיות במפרקים) והפרעות מטבוליות.
התקדמות המחלה אינה מהירה במיוחד, ראשית חולשת שרירים משפיעה על שרירי הרגליים, ולאחר מכן על הידיים. בדרך כלל, לחולים אין בעיות בבליעה ובתפקוד הנשימה.
רוב החולים עם SMA מסוג IV יכולים ללכת, ורק מעטים צריכים להיעזר בכסאות גלגלים.
SMA הקשור להפרה של הגן SMN נקרא SMA פרוקסימלי בספרות הרפואית - הם מהווים 95% מכלל האמיוטרופיות בעמוד השדרה. SMAs שאינם קשורים לגן SMN הם רבים למדי, אך הם נדירים. אלה כוללים, למשל, את מחלת קנדי. מחקר בשנות ה-90 הראה שמחלת קנדי אינה קשורה לפירוק של הגן SMN1, אלא למוטציות גנטיות אחרות המובילות לפגיעה בספיגה של חלבון SMN. המחלה מתבטאת באנשים מעל גיל 35. SMPA מאופיין בעיקר בחולשת גפיים.
סוג אחד של SMA שאינו קשור לגן SMN נקרא מחלת קנדי.העובדה שמחלה זו עדיין מכונה לפעמים SMA היא אנכרוניזם. בסוף שנות ה-60, כאשר ניוון זה תואר בפירוט, הוא נחשב לסוג של SMA, שכן הוא משפיע על אותם עצבים ושרירים כמו שלושת סוגי ה-SMA (אך במידה הרבה פחות).
איך זה מטופל?
נכון להיום, אין תרופה ל-SMA.
התאגיד הבינלאומי "ביוג'ן" פיתח את התרופה "ספינרזה", ששיפרה משמעותית את מצבם של חולים אליהם שימשה במהלך הבדיקה. נכון להיום התרופה מאושרת לשימוש בארה"ב, באירופה העלות המשוערת של קורס שנתי, על פי חישובי החברה, תעמוד על כ-270 אלף יורו, ברוסיה התרופה אינה מאושרת. טיפול לכל החיים.
האם אפשר לעזור לאנשים עם SMA ואיך בדיוק?
עדיין לא ניתן לרפא את המחלה, אך ניתן להקל על מצבם של חולי SMA, כלומר לפצות על ביטויי המחלה בדרכים שונות.
בסוגים חמורים של SMA, יש לעזור לאנשים לנשום ולבלוע. לכן, הם זקוקים באופן חיוני למאווררים ניידים, לשאובים, למכיחים, לשקיות אמבו.
גם ילדים עם SMA באמת זקוקים לעזרת מתנדבים שיכולים להחליף את הוריהם לפחות לזמן קצר.
ילדים עם SMA עשויים להזדקק לעזרה בכל עת, אז אמהות ואבות תמיד ערים ולומדים את מיומנויות ההחייאה הדרושות למקרה שהילד יפסיק לנשום פתאום.
חולים פחות קשים זקוקים לתרופות להקלת הנשימה, מחוכים, כסאות גלגלים ומכשירים אחרים המקלים על התנועה והחיים של אנשים עם שרירים חלשים.
מחלה שנמשכת שנים רבות היא מתישה, ולכן חולים, בעיקר מבוגרים, זקוקים לא פעם לעזרת פסיכולוג.

הקרן פועלת ברחבי רוסיה. לעבודת הקרן שני כיוונים עיקריים - מתן סיוע לחולי SMA עצמם וליקיריהם, ועבודה על שינויים מערכתיים במצב עם SMA ברוסיה.
תוכלו לתמוך בפעילות הקרן על ידי תרומה בכל דרך הנוחה לכם. ניתן לסייע בתרומה חד פעמית או רגילה בעמוד מיוחד של הקרן או בשליחת SMS למספר הקצר 3443 עם המילה SMA ובהפרדה ברווח, סכום התרומה - למשל, CMA 300.
האם ניתן לקבל SMA מחיסונים?
באירופה ובארצות הברית לא אותר הקשר בין חיסונים לביטוי המחלה.
כדי להבין אם יש קשר בין SMA לחיסונים עשוי להסביר את ההבדל בין SMA לפוליו. פולומיאליטיס היא מחלה זיהומית כאשר גופו של ילד בריא בתחילה ניזוק מזיהום. ילד עם SMA, שנולד עם גנום פגוע, עשוי להיראות בריא מבחוץ, אבל למעשה הוא כבר חולה, רק התסמינים של מחלתו מופיעים בהדרגה. בהקשר זה, SMA היא אותה מחלה "מושהית" כמו למשל, ניוון שרירים דושן או תסמונת רט, כאשר ילד המתפתח בהתאם לנורמה במשך זמן מה מאבד מיומנויות שנרכשו בעבר והופך לנכה.
רוב הביטויים של SMA קשורים לפיתוח המיומנויות המוטוריות הראשונות. הביטויים הראשונים של המחלה חופפים בזמן למספר חיסונים הקשורים לגיל. כתוצאה מכך, אדם וקרוביו עשויים לטעון כי הוא "חלה מהחיסון", אך למעשה הוא רק הראה סימני מחלה שכבר היו לו.
כיצד נקבע שלילד יש SMA ולא מחלה אחרת?
למרות העובדה ש-SMA תוארה לראשונה על ידי הנוירולוג האוסטרי גווידו ורדניג והנוירולוג הגרמני יוהאן הופמן בתחילת שנות ה-90, אופי המחלה הובן במלואו רק בסוף המאה ה-20. הגן SMN1 התגלה בשנת 1995. כדי לאשר את האבחנה של SMA, יש צורך בבדיקה גנטית.
ברוסיה, בדיקות גנטיות מתאימות הפכו לזמינות בתחילת שנות ה-2000. אפשר לעשות בדיקה גנטית ל-SMA לפי ביטוח בריאות חובה, אבל בפועל, לא יותר מדי רופאים מכירים את האבחנה הנדירה הזו ומפנים מטופלים למחקר מתאים. העלות של בדיקה כזו במעבדות מסחריות במוסקבה היא כ -6 אלף רובל.
היעדר אבחון ספציפי הוביל גם לבלבול באבחונים. רוב החולים ב-SMA ברוסיה לא זוהו, רבים מאלה שזוהו אובחנו עם מחלת ורדניג-הופמן, אם כי לא כולם (במיוחד מבוגרים) אכן חולים בסוג זה של מחלה.
כמה חולים עם SMA יש ברוסיה?

התרופה "Spinraza", אשר משפרת באופן משמעותי את מצב החולים. תמונה מאת healthbeat.spectrumhealth.org
בהתחשב בתדירות המחלה, מספר החולים ב-SMA ברוסיה צריך להיות בין שבעה לעשרים וארבעה אלף איש. נכון להיום, ישנם כ-400 איש בפנקס המטופלים של קרן משפחות SMA.
מי ברוסיה עוזר לאנשים עם SMA ולמשפחותיהם
קרן צדקה ורה, בית הוספיס לילדים עם מגדלור, קרן צדקה פליאטיבית לילדים, קרן צדקה משפחות SMA, שירות פליאטיבי לילדים רחמים.
משנת 2014 מתפתח במוסקבה פרויקט משותף לשירות "רחמים" וקרן "מרפאות SMA" "מרפאות SMA. במפגשים המתקיימים אחת לחודש יכולים המטופלים לקבל ייעוץ מרופא ריאות, אורטופד, פיזיותרפיסט ו פְּסִיכוֹלוֹג. לאחרונה חלק מהמפגשים מתמקדים בצרכי מטופלים מבוגרים.
אנשים מפורסמים עם SMA

סימון שפינוגליו האיטלקייהנולד עם מחלה תורשתית - ניוון שרירי עמוד השדרה סוג 2. היא לא מסוגלת ללכת מאז הלידה ומתנועעת רק בעזרת כסא גלגלים חשמלי. אבל חייה מלאים ועשירים; שום דבר לא יכול לעצור את הרצון שלה לחיות.
סימונה עובדת ב-Italian Famiglies of SMA, ומסייעת לילדים ומבוגרים עם SMA ומחלות עצב-שריר אחרות.
סימון גם הקליטה כמה שירים פופולריים בקהילת SMA האיטלקית - על החופש לעשות מה שאתה רוצה, למרות המחלה.
הזמרת הרוסייה יוליה סמוילובהנולדה באוכטה (רפובליקה קומי) בגיל עשר הופיעה בקונצרט צדקה, ולאחר מכן הוזמנה לשיר בארמון החלוצים המקומי. בגיל חמש עשרה החלה ללמוד בבית התרבות של העיר.
ב-2008 היא אספה להקה מוזיקלית משלה (פורקה ב-2010). בשנת 2013 היא השתתפה בתחרות פקטור A בערוץ הטלוויזיה רוסייה. היא זכתה במקום השני וקיבלה את הפרס האישי של אללה פוגצ'בה "כוכב הזהב של אללה". בשנת 2017, עקב אי קבלתה של רוסיה לתכנית התחרות, היא לא יכלה להשתתף באירוויזיון. נע בכיסא גלגלים.
מתכנת מוולדימיר ולרי ספירידונוב. הוא סיים את בית הספר עם מדליית זהב, ואז הגן על תואר ההנדסה שלו. בשנת 2015, ולרי תכננה להיות משתתפת בניסוי של המנתח האיטלקי סרג'יו קנוברו להשתלת ראש אנושי (הניסוי בוטל).
כיום ולרי הוא חבר בלשכה הציבורית של ולדימיר, מומחה לנושאי סביבה נגישה, וגם היוצר של קהילה משלו "תשוקה לחיים", שמדברת על יצירת סביבה נגישה ופרויקטים רפואיים מבטיחים. ולרי משתתפת בתוכניות טלוויזיה רבות בטלוויזיה הרוסית והזרה.